![]() Исследовано адсорбционное поведение вольфрамат-, изополи- и гетерополивольфрамат-анионов в 0,5 М ацетатном буферном растворе на границе раствор/ртуть и раствор/воздух. Анализ полученных данных свидетельствует о значительном специфическом взаимодействии вольфраматных анионов с поверхностью ртутного электрода. На основании предварительных оценок величин адсорбций катионов и анионов в области потенциалов адсорбции высказано предположение об образовании на поверхности ртути солеподобных адсорбционных слоев. Установлено, что в области потенциалов адсорбции вольфраматных анионов наблюдается сильное торможение реакции электровосстановления аниона BrO4–, аналогичное наблюдаемому в присутствии нейтральных органических веществ, образующих на поверхности электрода двумерные конденсированные слои.
Исследовано адсорбционное поведение вольфрамат-, изополи- и гетерополивольфрамат-анионов в 0,5 М ацетатном буферном растворе на границе раствор/ртуть и раствор/воздух. Анализ полученных данных свидетельствует о значительном специфическом взаимодействии вольфраматных анионов с поверхностью ртутного электрода. На основании предварительных оценок величин адсорбций катионов и анионов в области потенциалов адсорбции высказано предположение об образовании на поверхности ртути солеподобных адсорбционных слоев. Установлено, что в области потенциалов адсорбции вольфраматных анионов наблюдается сильное торможение реакции электровосстановления аниона BrO4–, аналогичное наблюдаемому в присутствии нейтральных органических веществ, образующих на поверхности электрода двумерные конденсированные слои.
![]() В присутствии вольфрамат-анионов (WO42–) реализуются разнообразные электрокаталитические процессы: выделение водорода [1], восстановление нитрат-, хлорат- и перхлорат-анионов [2,3] в кислых растворах. Высокой электрокаталитической активностью в различных реакциях обладают также гетерополисоединения и продукты их частичного восстановления (гетерополисини) [4–8]. Однако сложные адсорбционные явления, сопровождающие эти процессы, изучены лишь отрывочно [9–11]. Так, наблюдаемое в растворах NaClO4 резкое уменьшение периода капания ртутного капельного электрода, а также смещение максимума кривой зависимости периода капания от потенциала электрода при введении в раствор добавок Na2WO4, дали основание авторам [9,10] сделать вывод о значительной адсорбции вольфрамат-аниона.
В присутствии вольфрамат-анионов (WO42–) реализуются разнообразные электрокаталитические процессы: выделение водорода [1], восстановление нитрат-, хлорат- и перхлорат-анионов [2,3] в кислых растворах. Высокой электрокаталитической активностью в различных реакциях обладают также гетерополисоединения и продукты их частичного восстановления (гетерополисини) [4–8]. Однако сложные адсорбционные явления, сопровождающие эти процессы, изучены лишь отрывочно [9–11]. Так, наблюдаемое в растворах NaClO4 резкое уменьшение периода капания ртутного капельного электрода, а также смещение максимума кривой зависимости периода капания от потенциала электрода при введении в раствор добавок Na2WO4, дали основание авторам [9,10] сделать вывод о значительной адсорбции вольфрамат-аниона.
![]() В связи с попыткой исследования электрохимических превращений центрального иона гетерополисоединений как модельных процессов дальнего переноса электрона [12–15] вопрос о специфической адсорбции реагентов, определяющей их локализацию в реакционном слое, имеет первостепенное значение. В настоящей работе предпринята попытка моделирования адсорбционного поведения таких реагентов с использованием солей анионов WO42–, [H2W12O42]10–, [H2W12O40]6–, [PW12O40]3–, [CoW12O40]6–, электрохимически неактивных в водных растворах в широкой области потенциалов. Выбор этих изополи- и гетерополивольфраматов (далее - ИПВ и ГПВ) позволяет рассчитывать на выявление достаточно общих адсорбционных закономерностей: в широком интервале варьируются зарядность и структурные особенности, определяющие, в свою очередь, протоно-донорные свойства и устойчивость катион-анионных ассоциатов.
В связи с попыткой исследования электрохимических превращений центрального иона гетерополисоединений как модельных процессов дальнего переноса электрона [12–15] вопрос о специфической адсорбции реагентов, определяющей их локализацию в реакционном слое, имеет первостепенное значение. В настоящей работе предпринята попытка моделирования адсорбционного поведения таких реагентов с использованием солей анионов WO42–, [H2W12O42]10–, [H2W12O40]6–, [PW12O40]3–, [CoW12O40]6–, электрохимически неактивных в водных растворах в широкой области потенциалов. Выбор этих изополи- и гетерополивольфраматов (далее - ИПВ и ГПВ) позволяет рассчитывать на выявление достаточно общих адсорбционных закономерностей: в широком интервале варьируются зарядность и структурные особенности, определяющие, в свою очередь, протоно-донорные свойства и устойчивость катион-анионных ассоциатов.
![]() Электрокапиллярные кривые были получены как непосредственным измерением пограничного натяжения (s) на границе ртуть/раствор методом капиллярного электрометра Гуи, так и путем расчета s из величин периода капания ртутного капилляра (t) по формуле:
Электрокапиллярные кривые были получены как непосредственным измерением пограничного натяжения (s) на границе ртуть/раствор методом капиллярного электрометра Гуи, так и путем расчета s из величин периода капания ртутного капилляра (t) по формуле:
![]()
(m — скорость вытекания ртути из капилляра радиуса rk, t — период капания и g — ускорение силы тяжести) [16]. Необходимость непрямых измерений была вызвана тем, что в большинстве исследованных растворов адсорбция анионов приводила к прилипанию ртутного мениска, как, например, при адсорбции камфоры [17]. Анализ электрокапиллярных кривых, полученных двумя способами, показал, что прилипание ртути в капилляре в прямых измерениях приводит к завышенным величинам s, поэтому ниже мы приводим результаты расчета s из значений t.
![]() В качестве электролита фона использовался 0,5 М ацетатный буферный раствор (рН=4,7), потенциал (Е) измеряли относительно насыщенного каломельного электрода. Для измерения t,E-кривых, а также поляризационных (I,E-) кривых использовали капилляр со скоростью вытекания 6,5•10–4 г/с и временем жизни ртутной капли при разомкнутой цепи 10,1 с. Фиксировали мгновенные токи в конце жизни капли.
В качестве электролита фона использовался 0,5 М ацетатный буферный раствор (рН=4,7), потенциал (Е) измеряли относительно насыщенного каломельного электрода. Для измерения t,E-кривых, а также поляризационных (I,E-) кривых использовали капилляр со скоростью вытекания 6,5•10–4 г/с и временем жизни ртутной капли при разомкнутой цепи 10,1 с. Фиксировали мгновенные токи в конце жизни капли.
![]() Катодный предел исследуемой области потенциалов для анионов WO42–, ИПВ и ГПВ определялся потенциалами начала восстановления этих анионов (а также каталитического выделения водорода в их присутствии). Катодные токи, отвечающие этим процессам и превышающие токи заряжения, при рН 4,7 фиксировали в области Е < –0,9 В. Электрокапиллярные кривые в растворах с одинаковыми добавками ИПВ и ГПВ практически идентичны, репрезентативные s,Е-кривые приведены для растворов солей [H2W12O42]10–. Как видно из электрокапиллярных кривых (рис.1а,б), на положительно заряженной поверхности ртутного электрода (q>0) в присутствии вольфраматов наблюдается резкое понижение пограничного натяжения. Адсорбция исследованных анионов вызывает смещение потенциала нулевого заряда в отрицательную сторону, величина смещения зависит от объемной концентрации и достигает в исследуемом интервале ( 5•10–5 — 5•10–3 М WO42–; 10–5 — 10–3 М ИПВ и ГПВ) 120 мВ. Наблюдаемое снижение s существенно превышает снижение пограничного натяжения при переходе от раствора NaF к ацетатному буферному раствору.
Катодный предел исследуемой области потенциалов для анионов WO42–, ИПВ и ГПВ определялся потенциалами начала восстановления этих анионов (а также каталитического выделения водорода в их присутствии). Катодные токи, отвечающие этим процессам и превышающие токи заряжения, при рН 4,7 фиксировали в области Е < –0,9 В. Электрокапиллярные кривые в растворах с одинаковыми добавками ИПВ и ГПВ практически идентичны, репрезентативные s,Е-кривые приведены для растворов солей [H2W12O42]10–. Как видно из электрокапиллярных кривых (рис.1а,б), на положительно заряженной поверхности ртутного электрода (q>0) в присутствии вольфраматов наблюдается резкое понижение пограничного натяжения. Адсорбция исследованных анионов вызывает смещение потенциала нулевого заряда в отрицательную сторону, величина смещения зависит от объемной концентрации и достигает в исследуемом интервале ( 5•10–5 — 5•10–3 М WO42–; 10–5 — 10–3 М ИПВ и ГПВ) 120 мВ. Наблюдаемое снижение s существенно превышает снижение пограничного натяжения при переходе от раствора NaF к ацетатному буферному раствору.
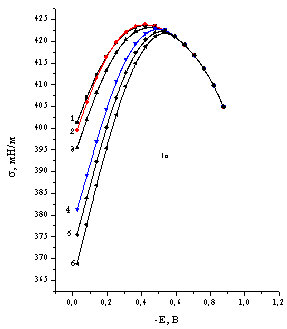 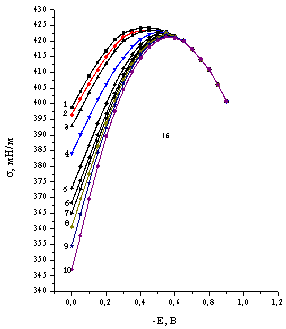 |
| Рис.1. Электрокапиллярные кривые, рассчитанные из величин периода капания ртутного электрода в 0,5 М ацетатных буферных растворах с добавками X мM Na2WO4 (а) и Y мМ Na10[H2W12O42] (б): X= 0(1), 0.05(2), 0.1(3), 0.5(4), 1(5), 3(6); Y= 0(1), 0.012(2), 0.015(3), 0.017(4), 0.025(5), 0.03(6), 0.05(7), 0.08(8), 0.1(9), 0.2(10). |
![]() Отметим, что эффекты понижения s в растворах нейтральных органических веществ каркасной структуры (камфора, борнеол) тех же концентраций выражены слабее, чем для исследованных нами вольфраматов. Указанные органические молекулы образуют особо прочные высоко упорядоченные адсорбционные слои - двумерные конденсированные слои (ДКС)[18–20].
Отметим, что эффекты понижения s в растворах нейтральных органических веществ каркасной структуры (камфора, борнеол) тех же концентраций выражены слабее, чем для исследованных нами вольфраматов. Указанные органические молекулы образуют особо прочные высоко упорядоченные адсорбционные слои - двумерные конденсированные слои (ДКС)[18–20].
![]() С целью выявления аналогий с адсорбционными явлениями для органических соединений методом наибольшего давления в пузырьке воздуха было измерено поверхностное натяжение на границе раствор/воздух в присутствии анионов WO42–, ИПВ и ГПВ. В указанных выше интервалах концентраций анионов поверхностное натяжение практически не отличалось от полученного в растворе фона. Сопоставление адсорбционного поведения анионов WO42–, ИПВ и ГПВ на границе раствор/воздух и на незаряженной поверхности ртутного электрода свидетельствует о достаточно сильном специфическом взаимодействии этих анионов с поверхностью ртути. По-видимому, основной вклад в хемосорбционные донорно-акцепторные взаимодействия сложной природы вносят концевые атомы кислорода оксовольфраматных октаэдров.
С целью выявления аналогий с адсорбционными явлениями для органических соединений методом наибольшего давления в пузырьке воздуха было измерено поверхностное натяжение на границе раствор/воздух в присутствии анионов WO42–, ИПВ и ГПВ. В указанных выше интервалах концентраций анионов поверхностное натяжение практически не отличалось от полученного в растворе фона. Сопоставление адсорбционного поведения анионов WO42–, ИПВ и ГПВ на границе раствор/воздух и на незаряженной поверхности ртутного электрода свидетельствует о достаточно сильном специфическом взаимодействии этих анионов с поверхностью ртути. По-видимому, основной вклад в хемосорбционные донорно-акцепторные взаимодействия сложной природы вносят концевые атомы кислорода оксовольфраматных октаэдров.
![]() Предварительные оценки показывают, что достигаемые заполнения поверхности ртутного электрода исследованными анионами близки к монослойным или даже превышают их в области потенциалов 0 — –0,5 В, причем формальный расчет в соответствии с формулами (10.1), (10.2) и (10.6) в [21] свидетельствует о том, что величины адсорбции катионов и анионов сопоставимы. Можно предположить, что в исследованных системах образуются солеподобные слои того же типа, что и на твердых поверхностях после адсорбции на них различных гетерополисоединений при разомкнутой цепи [22–25]. В [22,23] на основе туннельно-микроскопических исследований такие слои были квалифицированы как самоорганизованные. Более корректный количественный анализ может быть проведен в будущем по s,Е-кривым, полученным методом выдавливания ртутной капли из U-образного капилляра [26], позволяющим избежать прилипания ртути к стеклу.
Предварительные оценки показывают, что достигаемые заполнения поверхности ртутного электрода исследованными анионами близки к монослойным или даже превышают их в области потенциалов 0 — –0,5 В, причем формальный расчет в соответствии с формулами (10.1), (10.2) и (10.6) в [21] свидетельствует о том, что величины адсорбции катионов и анионов сопоставимы. Можно предположить, что в исследованных системах образуются солеподобные слои того же типа, что и на твердых поверхностях после адсорбции на них различных гетерополисоединений при разомкнутой цепи [22–25]. В [22,23] на основе туннельно-микроскопических исследований такие слои были квалифицированы как самоорганизованные. Более корректный количественный анализ может быть проведен в будущем по s,Е-кривым, полученным методом выдавливания ртутной капли из U-образного капилляра [26], позволяющим избежать прилипания ртути к стеклу.
![]() Выборочное полярографическое тестирование влияния сильно адсорбирующихся вольфраматов на кинетику электродных процессов на примере реакции электровосстановления аниона BrO4– в присутствии WO42– и [H2W12O42]10– показало, что при потенциалах положительнее –0,45 В имеет место сильное торможение указанной реакции (рис.2а,б). Аналогичный эффект наблюдался ранее в области потенциалов адсорбции органических веществ, образующих ДКС [27–29], и был объяснен в [28] перемещением зоны реакции на границу ДКС/раствор. При потенциале, близком к –0,45 В, наблюдается резкий ([H2W12O42]10–) или постепенный (WO42–) рост тока до значений, соответствующих току восстановления аниона BrO4– в отсутствие поверхностно-активных добавок. Отметим, что в [29] установлено влияние рН раствора на ширину области потенциалов торможения реакции восстановления аниона BrO4– в присутствии молекул органических веществ с не очень высоким значением рКа , формирующих на электроде ДКС. В то же время в растворах с добавками WO42– и [H2W12O42]10– отсутствует сколько-нибудь существенное смещение границ такой области при изменении рН раствора. Уменьшение концентрации катионов фона приводит к некоторому смещению потенциала резкого увеличения тока в присутствии анионов [H2W12O42]10– в сторону более положительных зарядов поверхности, что косвенно подтверждает предположение о существенной роли катионов в обсуждаемой картине адсорбционных явлений.
Выборочное полярографическое тестирование влияния сильно адсорбирующихся вольфраматов на кинетику электродных процессов на примере реакции электровосстановления аниона BrO4– в присутствии WO42– и [H2W12O42]10– показало, что при потенциалах положительнее –0,45 В имеет место сильное торможение указанной реакции (рис.2а,б). Аналогичный эффект наблюдался ранее в области потенциалов адсорбции органических веществ, образующих ДКС [27–29], и был объяснен в [28] перемещением зоны реакции на границу ДКС/раствор. При потенциале, близком к –0,45 В, наблюдается резкий ([H2W12O42]10–) или постепенный (WO42–) рост тока до значений, соответствующих току восстановления аниона BrO4– в отсутствие поверхностно-активных добавок. Отметим, что в [29] установлено влияние рН раствора на ширину области потенциалов торможения реакции восстановления аниона BrO4– в присутствии молекул органических веществ с не очень высоким значением рКа , формирующих на электроде ДКС. В то же время в растворах с добавками WO42– и [H2W12O42]10– отсутствует сколько-нибудь существенное смещение границ такой области при изменении рН раствора. Уменьшение концентрации катионов фона приводит к некоторому смещению потенциала резкого увеличения тока в присутствии анионов [H2W12O42]10– в сторону более положительных зарядов поверхности, что косвенно подтверждает предположение о существенной роли катионов в обсуждаемой картине адсорбционных явлений.
 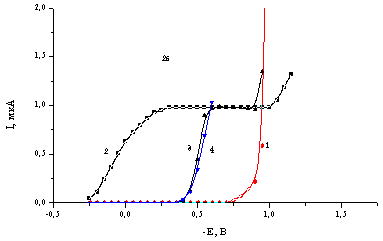 |
| Рис.2. Полярограммы восстановления, полученные на ртутном капельном электроде в растворах 0.5 М ацетатного буфера в присутствии (а): 5 мМ Na2WO4(1), 0.2 мМ LiBrO4(2), 0.2 мМ LiBrO4 с добавками 5 мМ Na2WO4 (3); (б): 0.5 мМ Na10[H2W12O42](1), 0.2 мМ LiBrO4(2), 0.2 мМ LiBrO4 с добавками 0.5 мМ Na10[H2W12O42](pH=4.7)(3), 0.2 мМ LiBrO4 с добавками 0.5 мМ Na10[H2W12O42](pH=2.5)(4). |
![]() Совокупность полученных данных позволяет предположить, что анионы WO42–, ИПВ и ГПВ склонны к самоорганизации и, возможно, двумерной конденсации на границе раствор/ртуть. Такое явление становится возможным благодаря ассоциации анионов с катионами раствора, стабилизирующей двумерный слой. Однако можно предположить, что более высокая диэлектрическая проницаемость адсорбционного неорганического слоя по сравнению с ДКС нейтральных органических молекул должна приводить, соответственно, к более высоким значениям дифференциальной емкости в области потенциалов адсорбции вольфраматных анионов по сравнению со значениями, характерными для органических веществ, образующих ДКС. Оценки, полученные путем двукратного дифференцирования электрокапиллярных кривых дают величины минимальных емкостей порядка 0,1 Ф/м2.
Совокупность полученных данных позволяет предположить, что анионы WO42–, ИПВ и ГПВ склонны к самоорганизации и, возможно, двумерной конденсации на границе раствор/ртуть. Такое явление становится возможным благодаря ассоциации анионов с катионами раствора, стабилизирующей двумерный слой. Однако можно предположить, что более высокая диэлектрическая проницаемость адсорбционного неорганического слоя по сравнению с ДКС нейтральных органических молекул должна приводить, соответственно, к более высоким значениям дифференциальной емкости в области потенциалов адсорбции вольфраматных анионов по сравнению со значениями, характерными для органических веществ, образующих ДКС. Оценки, полученные путем двукратного дифференцирования электрокапиллярных кривых дают величины минимальных емкостей порядка 0,1 Ф/м2.
![]() Вполне вероятно, что исследованные нами ранее процессы восстановления центрального иона в гетерополианионах [H6CoMo6O24]3– [13] и [MnW6O24]8– [12] при положительных зарядах поверхности протекают с участием прочно хемосорбированных реагентов. Разумеется, при переходе от вольфраматов к молибдатам (адсорбция которых не может быть исследована традиционными электрохимическими методами из-за более раннего, по сравнению с W(VI), восстановления Mo(VI)) количественные закономерности могут изменяться. Однако близость структурных свойств и состояния наружных кислородных атомов для этих классов гетерополисоединений позволяет ожидать аналогий адсорбционного поведения.
Вполне вероятно, что исследованные нами ранее процессы восстановления центрального иона в гетерополианионах [H6CoMo6O24]3– [13] и [MnW6O24]8– [12] при положительных зарядах поверхности протекают с участием прочно хемосорбированных реагентов. Разумеется, при переходе от вольфраматов к молибдатам (адсорбция которых не может быть исследована традиционными электрохимическими методами из-за более раннего, по сравнению с W(VI), восстановления Mo(VI)) количественные закономерности могут изменяться. Однако близость структурных свойств и состояния наружных кислородных атомов для этих классов гетерополисоединений позволяет ожидать аналогий адсорбционного поведения.
![]() Необходимо отметить, что вопрос о соотношении электростатической и хемосорбционной составляющих энергии адсорбции требует специального исследования. В частности, для гетерополианионов, содержащих некислые протоны в октаэдре гетероатома, может проявляться эффект ослабления адсорбции на положительно заряженной поверхности из-за значительного вклада отталкивания положительно заряженных и расположенных близко к поверхности протонов. Обнаруженное в настоящей работе явление резкого торможения электродного процесса вольфраматными адсорбатами позволяет рассчитывать на расширение круга систем для фиксации расстояния переноса электрона. В этом смысле вольфраматные адсорбционные слои могут, по-видимому, в будущем успешно конкурировать как с ДКС [18–20], так и с самоорганизованными алкантиольными монослоями [30].
Необходимо отметить, что вопрос о соотношении электростатической и хемосорбционной составляющих энергии адсорбции требует специального исследования. В частности, для гетерополианионов, содержащих некислые протоны в октаэдре гетероатома, может проявляться эффект ослабления адсорбции на положительно заряженной поверхности из-за значительного вклада отталкивания положительно заряженных и расположенных близко к поверхности протонов. Обнаруженное в настоящей работе явление резкого торможения электродного процесса вольфраматными адсорбатами позволяет рассчитывать на расширение круга систем для фиксации расстояния переноса электрона. В этом смысле вольфраматные адсорбционные слои могут, по-видимому, в будущем успешно конкурировать как с ДКС [18–20], так и с самоорганизованными алкантиольными монослоями [30].
![]() Авторы признательны проф. Б.Б.Дамаскину за ценные консультации. Работа выполнена в рамках проекта № 96–15–96995, поддержанного Президентом РФ.
Авторы признательны проф. Б.Б.Дамаскину за ценные консультации. Работа выполнена в рамках проекта № 96–15–96995, поддержанного Президентом РФ.