![]() The inhibition of various electrode reactions on mercury by adsorbed tungstate, WO42–, and dodecatungstate, [H2W12O42]10–, is reported for the region of positive electrode charges. For anionic reagents the degree of inhibition depends on the adsorption properties of reacting species and the value of a formal rate constant, whereas the reduction of Ag+ cation is found to remain unchanged in the same potential region. Kinetic data are considered jointly with the values of tungstates adsorption found from the decrease of surface tension. Possible constructions of reaction layers are compared, and the potential region in which continuous tungstate layer can be considered as a salt-like interlayer with barrier properties is singled out. A possibility to determine adsorption indirectly from kinetic data is also discussed.
The inhibition of various electrode reactions on mercury by adsorbed tungstate, WO42–, and dodecatungstate, [H2W12O42]10–, is reported for the region of positive electrode charges. For anionic reagents the degree of inhibition depends on the adsorption properties of reacting species and the value of a formal rate constant, whereas the reduction of Ag+ cation is found to remain unchanged in the same potential region. Kinetic data are considered jointly with the values of tungstates adsorption found from the decrease of surface tension. Possible constructions of reaction layers are compared, and the potential region in which continuous tungstate layer can be considered as a salt-like interlayer with barrier properties is singled out. A possibility to determine adsorption indirectly from kinetic data is also discussed.
![]() Long distance electron transfer (ET) presents a number of secondary phenomena important for molecular and biomolecular electronics. The problem of controllable operating modes for corresponding devices is not entirely solved, which stimulates the theoretical studies and its experimental verification for various model systems. In solution chemistry and electrochemistry, the exact reagent localization imposes most rigid restrictions on the systems of this sort, and a number of organic barrier layers on the electrodes were considered in this context during the last decade [1-3]. The disadvantage of well-studied alkanethiole layers [1] is defectiveness and pronounced electronic coupling with reagent, for condensed layers of organic steroids [2] the complications result from fast desorption at certain free charges of electrode, and the .most specific lipide-based layers [3] present a separate class of membrane-like films.
Long distance electron transfer (ET) presents a number of secondary phenomena important for molecular and biomolecular electronics. The problem of controllable operating modes for corresponding devices is not entirely solved, which stimulates the theoretical studies and its experimental verification for various model systems. In solution chemistry and electrochemistry, the exact reagent localization imposes most rigid restrictions on the systems of this sort, and a number of organic barrier layers on the electrodes were considered in this context during the last decade [1-3]. The disadvantage of well-studied alkanethiole layers [1] is defectiveness and pronounced electronic coupling with reagent, for condensed layers of organic steroids [2] the complications result from fast desorption at certain free charges of electrode, and the .most specific lipide-based layers [3] present a separate class of membrane-like films.
![]() Being quite stable at high free charges, the inorganic 2D-films with pronounced lateral interactions seem to be good candidates to supplement or, in some cases, serve in place of other barrier systems. Namely, tungstates and polytungstates demonstrate complete monolayer coverages on positively charged mercury at negligible bulk concentrations [4,5]. According to our recent preliminary note [6], this condensation results from coadsorption of multi-charged anions with supporting cations, and corresponding salt-like films are able to inhibite the electrode reactions.
Being quite stable at high free charges, the inorganic 2D-films with pronounced lateral interactions seem to be good candidates to supplement or, in some cases, serve in place of other barrier systems. Namely, tungstates and polytungstates demonstrate complete monolayer coverages on positively charged mercury at negligible bulk concentrations [4,5]. According to our recent preliminary note [6], this condensation results from coadsorption of multi-charged anions with supporting cations, and corresponding salt-like films are able to inhibite the electrode reactions.
![]() In this communication, we report the tungstate effects on a number of electrode reactions on mercury and present the assumptions concerning possible structures of reaction layers at tungstate-modified surfaces. The possibility to extract adsorption values from the data on ET kinetics is also considered.
In this communication, we report the tungstate effects on a number of electrode reactions on mercury and present the assumptions concerning possible structures of reaction layers at tungstate-modified surfaces. The possibility to extract adsorption values from the data on ET kinetics is also considered.
![]() Polarograms were measured on DME with the mercury flow rate of 0.65 mg/s and open circuit drop life of 10.1 s. Currents were recorded in the end of the drop life time. Potential E was measured and is given below in respect to SCE. For the supporting electrolyte, like in Ref.[6], a 0.5 M acetate buffer solution (pH 4.7) was used. All reagents were recrystallized from bidistilled water.
Polarograms were measured on DME with the mercury flow rate of 0.65 mg/s and open circuit drop life of 10.1 s. Currents were recorded in the end of the drop life time. Potential E was measured and is given below in respect to SCE. For the supporting electrolyte, like in Ref.[6], a 0.5 M acetate buffer solution (pH 4.7) was used. All reagents were recrystallized from bidistilled water.
![]() Quantitative study of adsorption equilibria was limited by slow diffusion in dilute solutions, and also by large molecular size preventing the use of conventional electrocapillary device. Below we are treating the data on surface tension obtained in Ref.[6] by means of the drop life measurements.
Quantitative study of adsorption equilibria was limited by slow diffusion in dilute solutions, and also by large molecular size preventing the use of conventional electrocapillary device. Below we are treating the data on surface tension obtained in Ref.[6] by means of the drop life measurements.
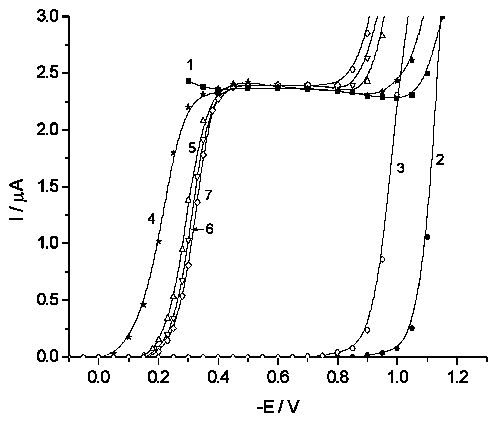
![]() Peroxodisulfate reduction on DME in acetate buffer medium proceeds under diffusion limitations up to E c.a. –1.1 V, at more negative potentials the proton discharge contribution becomes significant (Curve 1 in Fig.1). Tungstate-containing supporting electrolyte demonstrates no current response up to E c.a. –0.8 - –0.9 V (Curves 2,3 in Fig.1), the catalytic hydrogen evolution conjugated (or, probably, coupled) with tungstate reduction is beleived to start at more negative potentials.
Peroxodisulfate reduction on DME in acetate buffer medium proceeds under diffusion limitations up to E c.a. –1.1 V, at more negative potentials the proton discharge contribution becomes significant (Curve 1 in Fig.1). Tungstate-containing supporting electrolyte demonstrates no current response up to E c.a. –0.8 - –0.9 V (Curves 2,3 in Fig.1), the catalytic hydrogen evolution conjugated (or, probably, coupled) with tungstate reduction is beleived to start at more negative potentials.
 |
| Fig. 1. Polarograms of reduction measured on a dropping mercury electrode in 0.5 M acetate buffer (pH 4.7) in the presence of (1) 10–3 N K2SO4, (2) 5 10–3 M Na2WO4, (3) 2 10–4 M Na10[H2W12O42], (4) 10–3 N K2S2O8 with addition of (5–7) xM Na10[H2W12O42], x: (5) 2 10–4, (6) 5 10–4, (7) 10–3. |
![]() Polarografic maxima of the first kind, which prevents measuring at the potentials more positive than –0.3 V in tungstate-free solutions [7], never manifest themselves in the presence of tungstates (Curves 4–7 in Fig.1). In this case no current can be observed within the accuracy of c.a. 5 nA at potentials more positive than 0 and –0.15 V in the presence of WO42– and [H2W12O42]10–, respectively, and the limiting potential value (denoted below as E*) shifts towards more negative potentials with concentration of the additive (Curves 5–7 in Fig.1). The current coincides with the value for tungstate-free solution only starting from c.a. –0.4 V, in this region it demonstrates a pronounced dependence on the height of mercury column. At the same time, lower currents in the preceding region remain practically constant in these experiments, demonstrating their preferentially kinetic origin. Thus, we can be sure that the diffusion-layer structure does not change pronouncely in tungstate solutions, or, in other words, there are no adsorbate close-packed domains with large free areas between them.
Polarografic maxima of the first kind, which prevents measuring at the potentials more positive than –0.3 V in tungstate-free solutions [7], never manifest themselves in the presence of tungstates (Curves 4–7 in Fig.1). In this case no current can be observed within the accuracy of c.a. 5 nA at potentials more positive than 0 and –0.15 V in the presence of WO42– and [H2W12O42]10–, respectively, and the limiting potential value (denoted below as E*) shifts towards more negative potentials with concentration of the additive (Curves 5–7 in Fig.1). The current coincides with the value for tungstate-free solution only starting from c.a. –0.4 V, in this region it demonstrates a pronounced dependence on the height of mercury column. At the same time, lower currents in the preceding region remain practically constant in these experiments, demonstrating their preferentially kinetic origin. Thus, we can be sure that the diffusion-layer structure does not change pronouncely in tungstate solutions, or, in other words, there are no adsorbate close-packed domains with large free areas between them.
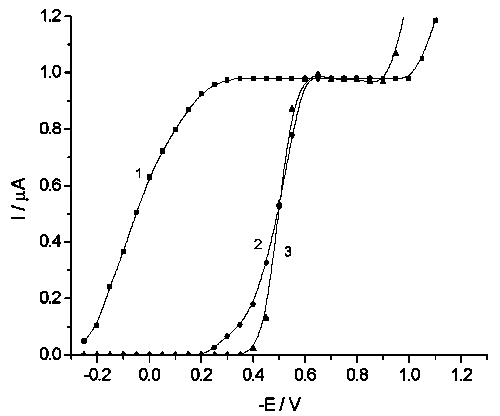
![]() Strong inhibition of bromate is also observed by both tungstate and dodecatungstate (Fig.2), with corresponding E* values of –0.2 and –0.35 V, respectively. The observed slopes of the waves (Curves 2, 3 in Fig.2) become lower in comparison with the usual wave (Curve 1, in Fig.2), manifesting pronounced desorption of tungstate in the narrow potential region between E* and the beginning of the region of limiting current.
Strong inhibition of bromate is also observed by both tungstate and dodecatungstate (Fig.2), with corresponding E* values of –0.2 and –0.35 V, respectively. The observed slopes of the waves (Curves 2, 3 in Fig.2) become lower in comparison with the usual wave (Curve 1, in Fig.2), manifesting pronounced desorption of tungstate in the narrow potential region between E* and the beginning of the region of limiting current.
 |
| Fig. 2. Polarograms of reduction measured on a dropping mercury electrode in 0.5 M acetate buffer (pH 4.7) in the presence of (1) 2*10–4 N LiBrO4, (2) with addition of 5 10–4 M Na2WO4, (3) with addition of 5 10–4 M Na10[H2W12O42]. |
![]() No dependence of E* on solution pH was found in the region of pH 2–5, in spite of pronounced pH-dependence of BrO4– reduction kinetics [12]. This fact confirms that the value of current in the region of tungstate adsorption is determined only by pH-independent coverage of the surface by the adsorbate under study.
No dependence of E* on solution pH was found in the region of pH 2–5, in spite of pronounced pH-dependence of BrO4– reduction kinetics [12]. This fact confirms that the value of current in the region of tungstate adsorption is determined only by pH-independent coverage of the surface by the adsorbate under study.
![]() Presented data on E* for both reactions agree qualitatively with comparative data on adsorption of tungstate and polytungstate species [6], the latter forming complete monolayer at lower concentrations and more negative potentials. In order to clarify the factors affecting the exact value of E*, we present below also the data for dodecatungstate and electrode processes with various mechanisms.
Presented data on E* for both reactions agree qualitatively with comparative data on adsorption of tungstate and polytungstate species [6], the latter forming complete monolayer at lower concentrations and more negative potentials. In order to clarify the factors affecting the exact value of E*, we present below also the data for dodecatungstate and electrode processes with various mechanisms.
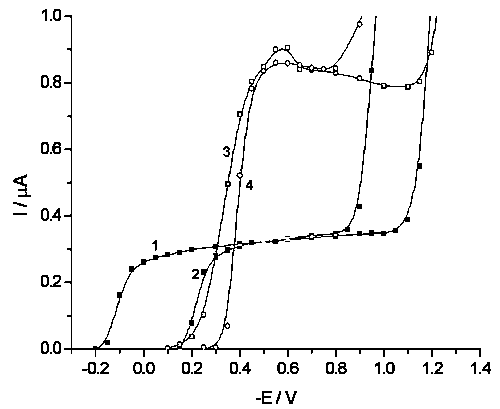
![]() A simple one-step ET without breaking of bond, [Co(III)EDTA]- reduction [8], also demonstrates the inhibition by dodecatungstate with a vanishingly small current at E > –0.15 V (see Curves 1 and 2 in Fig.3), and equal currents in the absence and in the presence of dodecatungstate at E < –0.4 V. There is no pronounced difference of E* for this and persulfate reactions, in spite of pronounced differnce of overvoltages (> 2 V) and, correspondingly, formal rate constants at given potential.
A simple one-step ET without breaking of bond, [Co(III)EDTA]- reduction [8], also demonstrates the inhibition by dodecatungstate with a vanishingly small current at E > –0.15 V (see Curves 1 and 2 in Fig.3), and equal currents in the absence and in the presence of dodecatungstate at E < –0.4 V. There is no pronounced difference of E* for this and persulfate reactions, in spite of pronounced differnce of overvoltages (> 2 V) and, correspondingly, formal rate constants at given potential.
 |
| Fig. 3. Polarograms of reduction measured on a dropping mercury electrode in 0.5 M acetate buffer (pH 4.7) in the presence of (1) 2*10–4 N Na[Co(edta), (2) 2 10–4 M Na3[NTSN], (3) with addition of 5*10–4 M Na10[H2W12O42]. |
![]() The multistep pH-dependent reduction of this reagent (denoted below as H3NTSN) is complicated by a number of chemical stages [9] and starts just in the vicinity of typical E*. Correspondingly, the inhibition is less pronounced than for other reagents (Curves 3 and 4 in Fig.3). The effect of tungstate disappears at about –0.6 V, the similar value is observed for pH-dependent BrO4– reduction (Fig.2), when for pH-independent ET no difference is observed already at –0.4 V. However, the lack of reliable mechanistic information prevents the unambiguous attributing of this feature to the proton participation in the limiting stage.
The multistep pH-dependent reduction of this reagent (denoted below as H3NTSN) is complicated by a number of chemical stages [9] and starts just in the vicinity of typical E*. Correspondingly, the inhibition is less pronounced than for other reagents (Curves 3 and 4 in Fig.3). The effect of tungstate disappears at about –0.6 V, the similar value is observed for pH-dependent BrO4– reduction (Fig.2), when for pH-independent ET no difference is observed already at –0.4 V. However, the lack of reliable mechanistic information prevents the unambiguous attributing of this feature to the proton participation in the limiting stage.
![]() It was mentioned in the literature that the reduction of cations, like Tl+, is not sensitive to the presence of various barrier layers [3]. The similar effect was observed by us for tungstate layers affecting Ag+ reduction; moreover, in contrast to results reported in [3], there was no evidence of even slight inhibition in the overall potential range.
It was mentioned in the literature that the reduction of cations, like Tl+, is not sensitive to the presence of various barrier layers [3]. The similar effect was observed by us for tungstate layers affecting Ag+ reduction; moreover, in contrast to results reported in [3], there was no evidence of even slight inhibition in the overall potential range.
![]() The most reliable explanation is coadsorption of cationic reagent with tungstate, i.e. its fast substitution for charge-compensating supporting cations. Another possible reason is the absence of any complications with the removal of product into solution bulk.
The most reliable explanation is coadsorption of cationic reagent with tungstate, i.e. its fast substitution for charge-compensating supporting cations. Another possible reason is the absence of any complications with the removal of product into solution bulk.
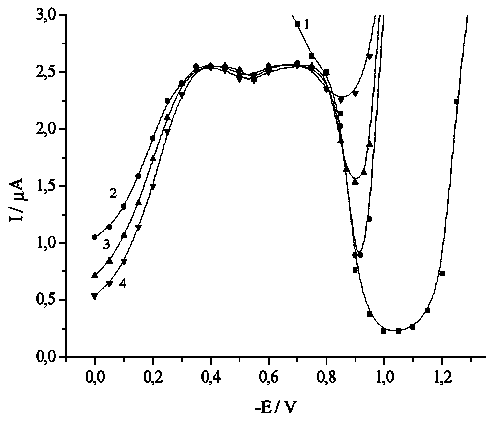
![]() If amalgam formation is accompanied by removal of products, like for reduction of tetrachloroplatinite, the inhibition takes place. In the region of acetate buffer stability, a pronounced current minima typical for this reaction (Curve 1 in Fig. 4) is observed, usually explained by electrostatic repulsion [7]. At positive mercury charges, the reduction in tungstate-free solutions occurs under diffusion control, but the measurements are complicated by polarographic maxima. The latter are suppressed by dodecatungstate (Curves 2–4 in Fig.4), and a limiting current is observed in the region from –0.3 to –0.8 V, with a small but well-reproducible pit in the vicinity of pzc (c.a. –0.5 V). This limiting current corresponds to the transfer of two electrons, and the diffusion coefficient calculated via Ilkovich equation equals 1.1*10–5 cm2/s, in good agreement with the values given in the literature [10]. Hence, we can conclude that the reduction of Pt(II) to Pt metal takes place. No complete inhibition is observed in the region under study.
If amalgam formation is accompanied by removal of products, like for reduction of tetrachloroplatinite, the inhibition takes place. In the region of acetate buffer stability, a pronounced current minima typical for this reaction (Curve 1 in Fig. 4) is observed, usually explained by electrostatic repulsion [7]. At positive mercury charges, the reduction in tungstate-free solutions occurs under diffusion control, but the measurements are complicated by polarographic maxima. The latter are suppressed by dodecatungstate (Curves 2–4 in Fig.4), and a limiting current is observed in the region from –0.3 to –0.8 V, with a small but well-reproducible pit in the vicinity of pzc (c.a. –0.5 V). This limiting current corresponds to the transfer of two electrons, and the diffusion coefficient calculated via Ilkovich equation equals 1.1*10–5 cm2/s, in good agreement with the values given in the literature [10]. Hence, we can conclude that the reduction of Pt(II) to Pt metal takes place. No complete inhibition is observed in the region under study.
 |
| Fig. 4. Polarograms of reduction measured on dropping mercury electrode in 0.5 M acetate buffer (pH 4.7) in the presence of 5*10–4 M K2PtCl4 with additions of xM Na10[H2W12O42], x: (1) 0, (2) 2*10–4, (3) 5*10–4, (4)*10–3. |
![]() We should stress that both S2O82– and PtCl42– reactions on mercury proceed at extremely high overvoltages in this potential region. For both anions the kinetic parameters of ET could be determined earlier only for the region of negative mercury charges, under strong inhibition by EDL field. When using tungstate interlayers, we are able to observe these anions reduction under kinetic mode also at positive electrode charges. When comparing the data for all reactions mentioned above, we can conclude that at least two factors can be, in general, responsible for degree of inhibition – the competitive specific adsorption of reacting anions (and/or reaction products) and tungstates, and also the value of a formal rate for certain reaction at potentials of tungstate adsorption.
We should stress that both S2O82– and PtCl42– reactions on mercury proceed at extremely high overvoltages in this potential region. For both anions the kinetic parameters of ET could be determined earlier only for the region of negative mercury charges, under strong inhibition by EDL field. When using tungstate interlayers, we are able to observe these anions reduction under kinetic mode also at positive electrode charges. When comparing the data for all reactions mentioned above, we can conclude that at least two factors can be, in general, responsible for degree of inhibition – the competitive specific adsorption of reacting anions (and/or reaction products) and tungstates, and also the value of a formal rate for certain reaction at potentials of tungstate adsorption.
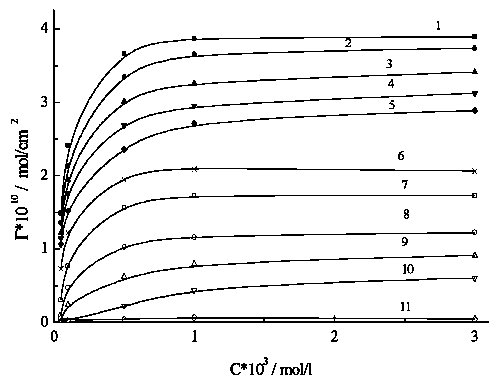
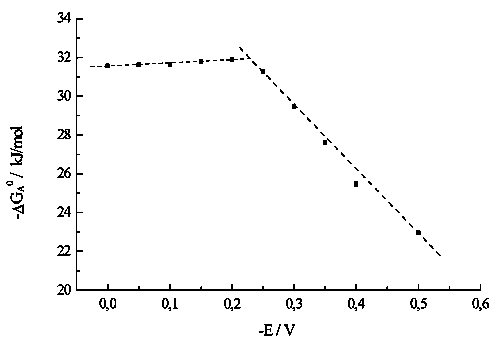
![]() This section presents a brief treatment of our data on surface tension obtained in [6], which was possible for tungstate adsorption only. Adsorption values reach the limiting potential-dependent values already in submillimolar range (Fig.5). Treating the curves in Fig.5 in the framework of Frumkin isotherm [11], we determined the adsorption equilibrium constant and calculated the value of adsorption Gibbs energy DGA, which can be considered qualitatively as a measure of surface activity. The main tendency observed for DGA vs E dependence (Fig.6) is a point of inflection at c.a.–0.2 V which can be attributed to some restructuring of adlayer. This characteristic value, in the first approximation, corresponds to E* for perbromate reduction (Fig.2). However, it is evidently more negative than the E* value for peroxodisulfate reduction (Fig.1).
This section presents a brief treatment of our data on surface tension obtained in [6], which was possible for tungstate adsorption only. Adsorption values reach the limiting potential-dependent values already in submillimolar range (Fig.5). Treating the curves in Fig.5 in the framework of Frumkin isotherm [11], we determined the adsorption equilibrium constant and calculated the value of adsorption Gibbs energy DGA, which can be considered qualitatively as a measure of surface activity. The main tendency observed for DGA vs E dependence (Fig.6) is a point of inflection at c.a.–0.2 V which can be attributed to some restructuring of adlayer. This characteristic value, in the first approximation, corresponds to E* for perbromate reduction (Fig.2). However, it is evidently more negative than the E* value for peroxodisulfate reduction (Fig.1).
 |
| Fig. 5. Isotherms of Na2WO4 adsorption from 0.5 M acetate-buffer solution (pH 4.7) on the mercury/solution interface at different potentials of mercury electrode, V: (1) 0, (2) –0.05, (3) –0.10, (4) –0.15, (5) –0.20, (6) –0.25, (7) –0.30, (8) –0.35, (9) –0.40, (10) –0.45, (11) –0.50. |
 |
| Fig. 6. Dependence of the adsorption energy of WO42- anions on the potential of mercury electrode in 0.5 M acetate-buffer solution (pH 4.7). |
![]() The portions of free surface, (1-q), can be obtained from G values (Fig.5) under assumption that the highest observed value (Gmax = 4*10-10 mol/cm2) corresponds to q
=1 (the corresponding square per adsorbed anion equals 0.42 nm2, the value close to the molecular size for solid tungstates). We used these values for rebuilding the Curve 4 in Fig.1 under an assumption that the S2O82- reduction occurs only on the free mercury surface, and ET via tungstate makes no contribution.
The portions of free surface, (1-q), can be obtained from G values (Fig.5) under assumption that the highest observed value (Gmax = 4*10-10 mol/cm2) corresponds to q
=1 (the corresponding square per adsorbed anion equals 0.42 nm2, the value close to the molecular size for solid tungstates). We used these values for rebuilding the Curve 4 in Fig.1 under an assumption that the S2O82- reduction occurs only on the free mercury surface, and ET via tungstate makes no contribution.
![]() Resulting values of currents (after correction for concentrational polarisation) appeared to depend slightly on WO42- concentration in solution bulk, confirming the competitive nature of tungstate and peroxodisulphate coadsorption. At the same time, these values are not very far from those ones extrapolated from currents reported in [7] for the region of negative mercury charges in concentrated solution, in the absence of pronounced EDL effects.
Resulting values of currents (after correction for concentrational polarisation) appeared to depend slightly on WO42- concentration in solution bulk, confirming the competitive nature of tungstate and peroxodisulphate coadsorption. At the same time, these values are not very far from those ones extrapolated from currents reported in [7] for the region of negative mercury charges in concentrated solution, in the absence of pronounced EDL effects.
![]() Hence, under rough approximation, non-zero currents observed in our experiments correspond preferentially to ET on the free electrode surface, and the difference of currents in the presence and absence of tungstates can be considered as a measure of adsorption.
Hence, under rough approximation, non-zero currents observed in our experiments correspond preferentially to ET on the free electrode surface, and the difference of currents in the presence and absence of tungstates can be considered as a measure of adsorption.
![]() We have applied the reverse procedure of treating polarograms for ET reactions of S2O82- and [Co(III)EDTA)]- inhibited by dodecatungstates in order to estimate the surface coverages by these polyanions, which can not be determined from surface tension data [6]. The resulting value of Gmax was found to be close to 1.9*10-10 mol/cm2, the corresponding square per adsorbed anion being 0.87 nm2. The narrow regions of adsorption isotherms constructed from available kinetic data were treated in terms of Frumkin isotherm. The most reliable results were obtained for E = -0.4 V, and gave the value of DGA = 42 kJ/mol (for tungstate, the adsorption energy equals 25.5 kJ/mol at the same potential).
We have applied the reverse procedure of treating polarograms for ET reactions of S2O82- and [Co(III)EDTA)]- inhibited by dodecatungstates in order to estimate the surface coverages by these polyanions, which can not be determined from surface tension data [6]. The resulting value of Gmax was found to be close to 1.9*10-10 mol/cm2, the corresponding square per adsorbed anion being 0.87 nm2. The narrow regions of adsorption isotherms constructed from available kinetic data were treated in terms of Frumkin isotherm. The most reliable results were obtained for E = -0.4 V, and gave the value of DGA = 42 kJ/mol (for tungstate, the adsorption energy equals 25.5 kJ/mol at the same potential).
![]() This result seems to be quite reasonable, as corresponding to a stronger adsorption of polytungstate. The sensitivity of ET current to tungstate adsorption is essentially higher than that of direct surface tension measurements, and thus presents an instrument of isotherm construction.
This result seems to be quite reasonable, as corresponding to a stronger adsorption of polytungstate. The sensitivity of ET current to tungstate adsorption is essentially higher than that of direct surface tension measurements, and thus presents an instrument of isotherm construction.
![]() A preliminary characterisation of tungstate barrier properties presented above clarifies several general features. First of all, in the presence of close-packed tungstate layer, the ET rate decreases by several orders of magnitude and becomes non-observable by conventional polarographic measurements. The cathodic limit of this strong inhibition region depends on the ability of a reagent to coadsorb with tungstate, namely, for cationic reagents complete inhibition is hardly possible due to their favorable coadsorption with tungstate anions; for anionic reagents, this limit is more negative, the stronger their adsorption ability.
A preliminary characterisation of tungstate barrier properties presented above clarifies several general features. First of all, in the presence of close-packed tungstate layer, the ET rate decreases by several orders of magnitude and becomes non-observable by conventional polarographic measurements. The cathodic limit of this strong inhibition region depends on the ability of a reagent to coadsorb with tungstate, namely, for cationic reagents complete inhibition is hardly possible due to their favorable coadsorption with tungstate anions; for anionic reagents, this limit is more negative, the stronger their adsorption ability.
![]() On the other hand, in the region of potentials, in which the layers are incomplete but the coverages are still high, ET takes place on the unoccupied electrode surface, most probably, at the isolated sites with molecular dimensions, and there is no evidence of pronounced decrease of current density per unoccupied surface area in comparison with this value in tungstate-free solution.This provides a sensitive instrument to determine the surface excess quantitatively from kinetic data.
On the other hand, in the region of potentials, in which the layers are incomplete but the coverages are still high, ET takes place on the unoccupied electrode surface, most probably, at the isolated sites with molecular dimensions, and there is no evidence of pronounced decrease of current density per unoccupied surface area in comparison with this value in tungstate-free solution.This provides a sensitive instrument to determine the surface excess quantitatively from kinetic data.
![]() The work is supported by the Russian Foundation of Basic Researches, project No 99-03-32367a.
The work is supported by the Russian Foundation of Basic Researches, project No 99-03-32367a.
![]()