![]() Впервые выполнено исследование динамического эффекта растворителя для процесса, протекающего в окрестности безактивационной области – реакции электровосстановления ансамбля аквахлоридных комплексов Pt(II) на отрицательно заряженном ртутном электроде. Проведен последовательный анализ влияния сахарозы на строение межфазной границы и энергии равновесной сольватации. Даны оценки указанных эффектов, необходимые для выделения зависимости скорости переноса электрона от времени релаксации растворителя из суммарной наблюдаемой зависимости от концентрации вязкообразующей добавки. Предложена методика оценки концентрации сахарозы в реакционном слое и приближенно учтено на этой основе увеличение локальной вязкости вблизи межфазной границы, обусловленное наличием поверхностного избытка сахарозы. Установлен интервал потенциалов, в котором исследуемая реакция протекает в режиме, близком к адиабатическому.
Впервые выполнено исследование динамического эффекта растворителя для процесса, протекающего в окрестности безактивационной области – реакции электровосстановления ансамбля аквахлоридных комплексов Pt(II) на отрицательно заряженном ртутном электроде. Проведен последовательный анализ влияния сахарозы на строение межфазной границы и энергии равновесной сольватации. Даны оценки указанных эффектов, необходимые для выделения зависимости скорости переноса электрона от времени релаксации растворителя из суммарной наблюдаемой зависимости от концентрации вязкообразующей добавки. Предложена методика оценки концентрации сахарозы в реакционном слое и приближенно учтено на этой основе увеличение локальной вязкости вблизи межфазной границы, обусловленное наличием поверхностного избытка сахарозы. Установлен интервал потенциалов, в котором исследуемая реакция протекает в режиме, близком к адиабатическому.
![]() Реакции электровосстановления анионов на отрицательно заряженном ртутном электроде долгое время служили удобными моделями для проверки представлений об электростатических взаимодействиях реагент-электрод [1]. Детальный анализ экспериментальных данных в рамках квантово-механической теории переноса электрона показал, что упомянутые реакции уникальны и в другом отношении: они дают возможность изучения специфики переноса электрона в окрестности безактивационной области, т.к. благодаря сильному электростатическому торможению электрохимической стадии диффузионных ограничений удается избежать при перенапряжениях до 2-3 В [2,3] в интервале шириной около 1 В.
Реакции электровосстановления анионов на отрицательно заряженном ртутном электроде долгое время служили удобными моделями для проверки представлений об электростатических взаимодействиях реагент-электрод [1]. Детальный анализ экспериментальных данных в рамках квантово-механической теории переноса электрона показал, что упомянутые реакции уникальны и в другом отношении: они дают возможность изучения специфики переноса электрона в окрестности безактивационной области, т.к. благодаря сильному электростатическому торможению электрохимической стадии диффузионных ограничений удается избежать при перенапряжениях до 2-3 В [2,3] в интервале шириной около 1 В.
![]() Одним из классических процессов обсуждаемого типа является восстановление аквахлоридных комплексов Pt(II) на отрицательно заряженном капающем ртутном электроде [4-9] (доступный интервал перенапряжений – от 1,2 до 2,3 В. В отличие от реагентов, рассмотренных в [2,3] (S2O82- и Fe(CN)63-), для Pt(II) минимум тока восстановления не исчезает при повышении концентрации электролита фона (максимальная исследованная концентрация составляла 3.5 М, NaCl). Ранее этот эффект связывали с адсорбцией реагента [4], взаимодействием Pt(II) с ртутью [5], ускорением процесса гидролиза вблизи электрода [9], однако однозначного количественного объяснения найдено не было.
Одним из классических процессов обсуждаемого типа является восстановление аквахлоридных комплексов Pt(II) на отрицательно заряженном капающем ртутном электроде [4-9] (доступный интервал перенапряжений – от 1,2 до 2,3 В. В отличие от реагентов, рассмотренных в [2,3] (S2O82- и Fe(CN)63-), для Pt(II) минимум тока восстановления не исчезает при повышении концентрации электролита фона (максимальная исследованная концентрация составляла 3.5 М, NaCl). Ранее этот эффект связывали с адсорбцией реагента [4], взаимодействием Pt(II) с ртутью [5], ускорением процесса гидролиза вблизи электрода [9], однако однозначного количественного объяснения найдено не было.
![]() Моделирование кинетики восстановления анионов в [2,3] проводили в рамках неадиабатического приближения (слабое взаимодействие электрод-реагент) [10,11]. Возможность распространения этого подхода на случай реакции восстановления анионных комплексов Pt(II), с учетом недавних результатов квантовохимического моделирования систем Pt(II)-Hg [12], не вполне очевидна. Есть основания предполагать, что перенос электрона в обсуждаемой системе, по крайней мере в некотором интервале потенциалов, является адиабатическим. В этом случае следует ожидать зависимости его скорости от релаксационных свойств среды (динамический эффект растворителя).
Моделирование кинетики восстановления анионов в [2,3] проводили в рамках неадиабатического приближения (слабое взаимодействие электрод-реагент) [10,11]. Возможность распространения этого подхода на случай реакции восстановления анионных комплексов Pt(II), с учетом недавних результатов квантовохимического моделирования систем Pt(II)-Hg [12], не вполне очевидна. Есть основания предполагать, что перенос электрона в обсуждаемой системе, по крайней мере в некотором интервале потенциалов, является адиабатическим. В этом случае следует ожидать зависимости его скорости от релаксационных свойств среды (динамический эффект растворителя).
![]() Для экспериментального исследования динамического эффекта растворителя в настоящей работе выбран подход, ранее апробированный для водных растворов [13-16] и основанный на установлении зависимости тока от вязкости раствора
Для экспериментального исследования динамического эффекта растворителя в настоящей работе выбран подход, ранее апробированный для водных растворов [13-16] и основанный на установлении зависимости тока от вязкости раствора ![]() , варьируемой путем введения добавок вязкообразователя [13–16]. Наиболее распространенный альтернативный подход – сравнение токов в разных растворителях (см. обзоры [17-19]) – в данном случае не имеет смысла, т.к. не обеспечивает постоянства состава частиц реагентов (аквакомплексов). Определение зависимости тока от давления (см, например, [20]) значительно сложнее в аппаратурном плане и, как и другие подходы, не всегда однозначно в смысле интерпретации.
, варьируемой путем введения добавок вязкообразователя [13–16]. Наиболее распространенный альтернативный подход – сравнение токов в разных растворителях (см. обзоры [17-19]) – в данном случае не имеет смысла, т.к. не обеспечивает постоянства состава частиц реагентов (аквакомплексов). Определение зависимости тока от давления (см, например, [20]) значительно сложнее в аппаратурном плане и, как и другие подходы, не всегда однозначно в смысле интерпретации.
![]() В качестве вязкообразователя использовали сахарозу С12Н22О11 (a-D-глюкопиранозил-b-D-фруктофуранозид), которая не восстанавливается на ртутном электроде в достаточно широком интервале потенциалов [21,22] (в отличие от менее инертной глюкозы [14]). Очевидно, что введение добавок сахарозы может приводить к изменению не только релаксационных, но и других свойств реакционной системы.
В качестве вязкообразователя использовали сахарозу С12Н22О11 (a-D-глюкопиранозил-b-D-фруктофуранозид), которая не восстанавливается на ртутном электроде в достаточно широком интервале потенциалов [21,22] (в отличие от менее инертной глюкозы [14]). Очевидно, что введение добавок сахарозы может приводить к изменению не только релаксационных, но и других свойств реакционной системы.
| (1) |
![]() В настоящей работе получены зависимости тока восстановления Рt(II) от концентрации сахарозы c(сах) и выполнен их всесторонний анализ. При этом предполагали, что строение реакционного слоя при изменении c(сах), по крайней мере на свободных от адсорбированной сахарозы участках, остается неизменным. Косвенным аргументом в пользу такого предположения служат результаты [21], указывающие на слабые изменения адсорбционного поведения хлорид-иона и сахарозы в условиях их соадсорбции. Поскольку закономерности адсорбции аквахлоридных комплексов Pt(II) и хлорид-ионов близки, можно рассчитывать на аналогичную ситуацию и в исследуемой системе.
В настоящей работе получены зависимости тока восстановления Рt(II) от концентрации сахарозы c(сах) и выполнен их всесторонний анализ. При этом предполагали, что строение реакционного слоя при изменении c(сах), по крайней мере на свободных от адсорбированной сахарозы участках, остается неизменным. Косвенным аргументом в пользу такого предположения служат результаты [21], указывающие на слабые изменения адсорбционного поведения хлорид-иона и сахарозы в условиях их соадсорбции. Поскольку закономерности адсорбции аквахлоридных комплексов Pt(II) и хлорид-ионов близки, можно рассчитывать на аналогичную ситуацию и в исследуемой системе.
![]() Методика полярографического эксперимента была такой же, как в [8], параметры капилляра составляли: скорость вытекания ртути m = 6.93·10-7 мг/с, период капания t = 11 с. Поправку на ток заряжения вносили по данным для соответствующих растворов КСl с добавками сахарозы. Токи восстановления сахарозы даже при высоких (до 2 М) концентрациях были пренебрежимо малы при Е > –1,6¸–1,8 В (нас. к. э.) (Здесь и далее все значения потенциалов Е приведены в шкале насыщенного каломельного электрода.).
Методика полярографического эксперимента была такой же, как в [8], параметры капилляра составляли: скорость вытекания ртути m = 6.93·10-7 мг/с, период капания t = 11 с. Поправку на ток заряжения вносили по данным для соответствующих растворов КСl с добавками сахарозы. Токи восстановления сахарозы даже при высоких (до 2 М) концентрациях были пренебрежимо малы при Е > –1,6¸–1,8 В (нас. к. э.) (Здесь и далее все значения потенциалов Е приведены в шкале насыщенного каломельного электрода.).
![]() Воду, соли и аргон, применяемый для деаэрирования, очищали как описано в [8]. Сахарозу марки MERCK extra pure использовали без дополнительной очистки. Рабочие растворы готовили непосредственно перед началом эксперимента разбавлением более концентрированного раствора K2PtCl4, приготовленного не более чем за несколько часов до измерений и хранившегося при температуре 0 – 5 оС. Степень гидролиза PtCl42-, достигавшаяся в рабочих растворах в ходе измерений, была близка к степени гидролиза в свежеприготовленных растворах [8].
Воду, соли и аргон, применяемый для деаэрирования, очищали как описано в [8]. Сахарозу марки MERCK extra pure использовали без дополнительной очистки. Рабочие растворы готовили непосредственно перед началом эксперимента разбавлением более концентрированного раствора K2PtCl4, приготовленного не более чем за несколько часов до измерений и хранившегося при температуре 0 – 5 оС. Степень гидролиза PtCl42-, достигавшаяся в рабочих растворах в ходе измерений, была близка к степени гидролиза в свежеприготовленных растворах [8].
![]() Коэффициенты диффузии D рассчитывали из предельных диффузионных токов Id по формуле Ильковича:
Коэффициенты диффузии D рассчитывали из предельных диффузионных токов Id по формуле Ильковича:
| (2) |
где n = 2 – число электронов, участвующих в реакции; [с] – концентрация реагента, численный коэффициент отвечает системе СИ.
![]() Относительную вязкость
Относительную вязкость ![]() растворов определяли по эмпирической формуле [25]
растворов определяли по эмпирической формуле [25]
| (3) |
которая в исследуемом интервале концентраций удовлетворительно согласуется с более поздними данными [26]. Согласно [26], вязкость растворов сахарозы при введении добавок NaСl в концентрации вплоть до 1 М изменяется незначительно (не более чем на несколько процентов), поэтому соответствующую поправку к формуле (3) не вводили.
![]() Значение
Значение ![]() в объеме использованных растворов 1–6 с концентрацией сахарозы c(сах) приведены в таблице, в ней также даны оптические и статические диэлектрические проницаемости (по данным, цитируемым в [14]), отвечающие им пекаровские факторы (рассчитанные по уравнению (1)) и времена релаксации растворителя.
в объеме использованных растворов 1–6 с концентрацией сахарозы c(сах) приведены в таблице, в ней также даны оптические и статические диэлектрические проницаемости (по данным, цитируемым в [14]), отвечающие им пекаровские факторы (рассчитанные по уравнению (1)) и времена релаксации растворителя.
Таблица 1. Характеристики исследуемых растворов.
| |||||||||||||||||||||||||||||||||||||||||||||||||
| * См. ниже |
![]() В тестовых экспериментах из предельных диффузионных токов восстановления Tl+ и Fe(CN)63- были рассчитаны коэффициенты диффузии D в исследуемых растворах сахарозы. В случае Fe(CN)63- наблюдалась линейная зависимость D от
В тестовых экспериментах из предельных диффузионных токов восстановления Tl+ и Fe(CN)63- были рассчитаны коэффициенты диффузии D в исследуемых растворах сахарозы. В случае Fe(CN)63- наблюдалась линейная зависимость D от ![]() , в то время как для Tl+ коэффициенты диффузии снижались с ростом с(сах) более резко. Последний результат интерпретировали как следствие образования комплекса катиона Tl+ с сахарозой. Согласно [27,28], обязательным стерическим требованием для комплексообразования с сахарозой является не слишком малый ионный радиус катиона металла (>0.08 нм). По данным, приведенным в [29], атомные радиусы Tl+ и Pt2+ равны соответственно 0.149 и 0.085 нм, что позволяет сделать предварительный вывод о том, что образование комплексов Pt(II) с сахарозой маловероятно. Косвенно этот вывод подтверждается данными [30] об отсутствии образования комплексных интермедиатов при восстановлении солей платины различными сахарами в щелочной среде. Для независимого контроля состояния Pt(II) измеряли cпектры поглощения рабочих растворов K2PtCl4 в диапазоне 280 – 500 нм при помощи спектрофотометра Unicam SP1800, используя в качестве растворов сравнения соответствующие фоновые растворы хлорида калия и сахарозы. Полученные спектры практически не отличались от измеренных ранее спектров в растворах, не содержащих сахарозы [8], обнаруживая слабую и несистематическую зависимость от концентрации добавки. Таким образом, нет оснований предполагать какие-либо осложнения в поведении исследуемых систем в результате комплексообразования, и можно рассчитывать на сохранение соотношения разных форм аквахлоридных комплексов во всей обсуждаемой серии растворов.
, в то время как для Tl+ коэффициенты диффузии снижались с ростом с(сах) более резко. Последний результат интерпретировали как следствие образования комплекса катиона Tl+ с сахарозой. Согласно [27,28], обязательным стерическим требованием для комплексообразования с сахарозой является не слишком малый ионный радиус катиона металла (>0.08 нм). По данным, приведенным в [29], атомные радиусы Tl+ и Pt2+ равны соответственно 0.149 и 0.085 нм, что позволяет сделать предварительный вывод о том, что образование комплексов Pt(II) с сахарозой маловероятно. Косвенно этот вывод подтверждается данными [30] об отсутствии образования комплексных интермедиатов при восстановлении солей платины различными сахарами в щелочной среде. Для независимого контроля состояния Pt(II) измеряли cпектры поглощения рабочих растворов K2PtCl4 в диапазоне 280 – 500 нм при помощи спектрофотометра Unicam SP1800, используя в качестве растворов сравнения соответствующие фоновые растворы хлорида калия и сахарозы. Полученные спектры практически не отличались от измеренных ранее спектров в растворах, не содержащих сахарозы [8], обнаруживая слабую и несистематическую зависимость от концентрации добавки. Таким образом, нет оснований предполагать какие-либо осложнения в поведении исследуемых систем в результате комплексообразования, и можно рассчитывать на сохранение соотношения разных форм аквахлоридных комплексов во всей обсуждаемой серии растворов.
![]() Поляризационные кривые в серии растворов 5·10-4 M K2PtCl4 с электролитом фона KCl и добавками сахарозы (табл. 1) представлены на рис. 1а - в. Общий вид кривых с минимумами, наблюдаемыми даже при высоких концентрациях электролита фона, аналогичен описанному ранее для обычных водных растворов [4–9].
Поляризационные кривые в серии растворов 5·10-4 M K2PtCl4 с электролитом фона KCl и добавками сахарозы (табл. 1) представлены на рис. 1а - в. Общий вид кривых с минимумами, наблюдаемыми даже при высоких концентрациях электролита фона, аналогичен описанному ранее для обычных водных растворов [4–9].
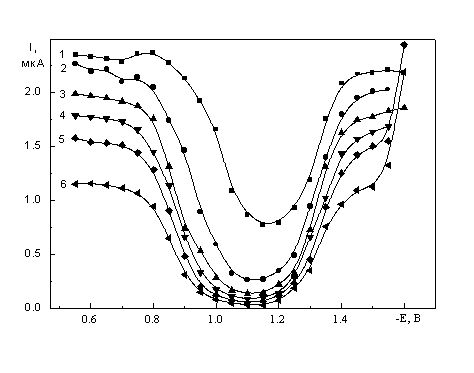 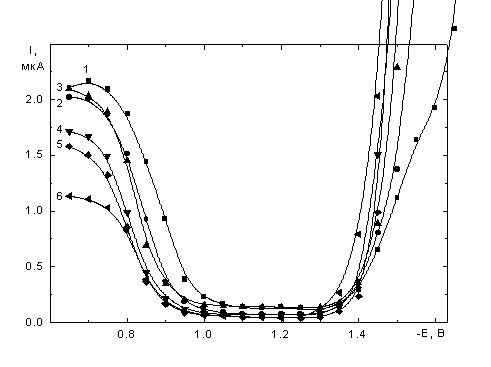 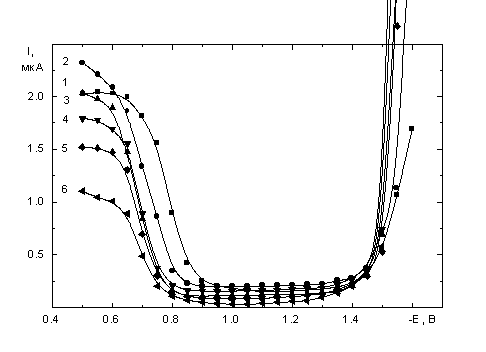 |
| Рис. 1. Поляризационные кривые растворов 5·10-4 M K2PtCl4 + x M KCl + y M сахароза, x: а - 1, б - 0.1, в - 0.01; y: 1 – 0, 2 – 0.117, 3 – 0.234, 4 – 0.468, 5 – 0.701, 6 – 1.17. |
![]() На полярограммах наблюдается максимум в области потенциала нулевого заряда (на рис. 1 не приведен). С увеличением c(сах) высота максимума уменьшается, а при c(сах) > 1 М максимум полностью исчезает, что связано, по-видимому, с адсорбцией сахарозы (при потенциалах вблизи точки нулевого заряда при не слишком малых концентрациях достигаются заполнения поверхности сахарозой более 0.9 [21,22]).
На полярограммах наблюдается максимум в области потенциала нулевого заряда (на рис. 1 не приведен). С увеличением c(сах) высота максимума уменьшается, а при c(сах) > 1 М максимум полностью исчезает, что связано, по-видимому, с адсорбцией сахарозы (при потенциалах вблизи точки нулевого заряда при не слишком малых концентрациях достигаются заполнения поверхности сахарозой более 0.9 [21,22]).
![]() Снижение тока при увеличении c(сах) наблюдается как в области предельного диффузионного тока, так и в области минимума. Форма и взаимное расположение полярографических кривых на участке после области минимума различаются для концентрации фона 1 M (рис. 1а) и более разбавленных по KCl растворов (рис. 1 б, в). Кривые для растворов без добавок сахарозы (кривые 1 на рис. 1 а–в) по величинам тока в минимуме сходны с кривыми [8] для свежеприготовленных растворов: в случае 1 M KCl при высоких отрицательных потенциалах наблюдается небольшая площадка (предположительно предельный диффузионный ток), а в более разбавленных по КСl растворах она отсутствует. На восходящих ветвях кривых рост тока до значений выше предельного диффузионного (по Рt(II)) начинается при потенциалах тем более отрицательных, чем ниже концентрация электролита фона. Природа участка резкого роста тока специально не исследовалась, можно предположить, что он обусловлен выделением водорода из внутрисферной воды в составе аквахлоридных комплексов, объемная концентрация которых тем выше, чем ниже концентрация КСl.
Снижение тока при увеличении c(сах) наблюдается как в области предельного диффузионного тока, так и в области минимума. Форма и взаимное расположение полярографических кривых на участке после области минимума различаются для концентрации фона 1 M (рис. 1а) и более разбавленных по KCl растворов (рис. 1 б, в). Кривые для растворов без добавок сахарозы (кривые 1 на рис. 1 а–в) по величинам тока в минимуме сходны с кривыми [8] для свежеприготовленных растворов: в случае 1 M KCl при высоких отрицательных потенциалах наблюдается небольшая площадка (предположительно предельный диффузионный ток), а в более разбавленных по КСl растворах она отсутствует. На восходящих ветвях кривых рост тока до значений выше предельного диффузионного (по Рt(II)) начинается при потенциалах тем более отрицательных, чем ниже концентрация электролита фона. Природа участка резкого роста тока специально не исследовалась, можно предположить, что он обусловлен выделением водорода из внутрисферной воды в составе аквахлоридных комплексов, объемная концентрация которых тем выше, чем ниже концентрация КСl.
![]() Вопрос о природе тока в области минимума имеет для задач настоящей работы принципиальное значение, поскольку при анализе динамического эффекта растворителя в элементарном акте переноса электрона необходимо исключить возможные вклады диффузионных ограничений. В условиях восстановления ансамбля реагентов нельзя исключить ситуации протекания одного из параллельных процессов восстановления на предельном диффузионном токе, который из-за одновременного восстановления других видов частиц не обнаруживается по характерной площадке. Ранее в [5] указывалось на то, что в области минимума в водных растворах лимитирующей стадией восстановления является переноса электрона, однако этого вывод нуждается в дополнительном подтверждении для растворов, содержащих сахарозу.
Вопрос о природе тока в области минимума имеет для задач настоящей работы принципиальное значение, поскольку при анализе динамического эффекта растворителя в элементарном акте переноса электрона необходимо исключить возможные вклады диффузионных ограничений. В условиях восстановления ансамбля реагентов нельзя исключить ситуации протекания одного из параллельных процессов восстановления на предельном диффузионном токе, который из-за одновременного восстановления других видов частиц не обнаруживается по характерной площадке. Ранее в [5] указывалось на то, что в области минимума в водных растворах лимитирующей стадией восстановления является переноса электрона, однако этого вывод нуждается в дополнительном подтверждении для растворов, содержащих сахарозу.
![]() Для выявления возможных диффузионных ограничений были экспериментально исследованы зависимости тока I от высоты ртутного столба h при разных потенциалах электрода E. Примеры полученных I,
Для выявления возможных диффузионных ограничений были экспериментально исследованы зависимости тока I от высоты ртутного столба h при разных потенциалах электрода E. Примеры полученных I, ![]() зависимостей приведены на рис. 2. Результаты анализировали, сопоставляя параметры A и B интерполированной зависимости
зависимостей приведены на рис. 2. Результаты анализировали, сопоставляя параметры A и B интерполированной зависимости ![]() . При потенциалах положительнее –0.7 В коэффициент A близок к нулю, при смешении потенциала в сторону более отрицательных значений он увеличивается, при переходе к области минимума резко снижается до значений, близких к I. Одновременно коэффициент B медленно уменьшается до значения, близкого к нулю. Это указывает на плавный переход от диффузионных ограничений к смешанному, а затем к кинетическому режиму, в том числе доказывает, что в области минимума диффузионные ограничения практически отсутствуют. Таким образом, ток в области минимума можно использовать для анализа динамического эффекта растворителя.
. При потенциалах положительнее –0.7 В коэффициент A близок к нулю, при смешении потенциала в сторону более отрицательных значений он увеличивается, при переходе к области минимума резко снижается до значений, близких к I. Одновременно коэффициент B медленно уменьшается до значения, близкого к нулю. Это указывает на плавный переход от диффузионных ограничений к смешанному, а затем к кинетическому режиму, в том числе доказывает, что в области минимума диффузионные ограничения практически отсутствуют. Таким образом, ток в области минимума можно использовать для анализа динамического эффекта растворителя.
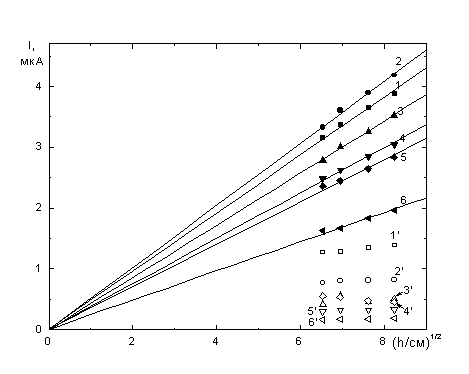 |
| Рис. 2. Зависимость от высоты столба ртути тока восстановления 5·10-4 M K2PtCl4 + 1 M KCl + x M сахарозы, x: (1,1’) – 0, (2,2’) – 0.117, (3,3) - 0.234, (4,4’) - 0.468, (5,5’) - 0.701, (6,6’) – 1.17 при потенциалах –0.7 (1 – 6) и –1.1 (1’ – 6’) В. |
![]() Коэффициенты диффузии D рассчитывали из величин Id (2), определенных в области строгой пропорциональности тока величине h1/2. Зависимости D от обратной вязкости
Коэффициенты диффузии D рассчитывали из величин Id (2), определенных в области строгой пропорциональности тока величине h1/2. Зависимости D от обратной вязкости ![]() представлены на рис. 3, они удовлетворительно описываются прямой линией при c(сах) > 0.2 M. Как наблюдалось ранее и в [8], с увеличением c(KCl) при одинаковой степени старения раствора D возрастает. Некоторое отклонение D,
представлены на рис. 3, они удовлетворительно описываются прямой линией при c(сах) > 0.2 M. Как наблюдалось ранее и в [8], с увеличением c(KCl) при одинаковой степени старения раствора D возрастает. Некоторое отклонение D, ![]() –зависимостей от прямой при малых c(сах) может быть связано с недостаточной точностью определения Id по кривым с наименее выраженными площадками. Отметим, что в условиях участия в реакции ансамбля комплексов предельный диффузионный ток является суммой предельных диффузионных токов по каждой из форм реагента, а рассчитанные по уравнению Ильковича величины D следует рассматривать как эффективные коэффициенты диффузии. Различия длины и формы площадок предельного тока в растворах с разными концентрациями сахарозы можно объяснить специфичными зависимостями скоростей каждого из параллельных процессов от вязкости, что, в свою очередь, может повлиять на потенциал выхода каждого из них на соответствующий парциальный предельный ток. В целом найденные зависимости D от обратной вязкости согласуются с предположением о неизменности состояния реагента в растворах изучаемой серии.
–зависимостей от прямой при малых c(сах) может быть связано с недостаточной точностью определения Id по кривым с наименее выраженными площадками. Отметим, что в условиях участия в реакции ансамбля комплексов предельный диффузионный ток является суммой предельных диффузионных токов по каждой из форм реагента, а рассчитанные по уравнению Ильковича величины D следует рассматривать как эффективные коэффициенты диффузии. Различия длины и формы площадок предельного тока в растворах с разными концентрациями сахарозы можно объяснить специфичными зависимостями скоростей каждого из параллельных процессов от вязкости, что, в свою очередь, может повлиять на потенциал выхода каждого из них на соответствующий парциальный предельный ток. В целом найденные зависимости D от обратной вязкости согласуются с предположением о неизменности состояния реагента в растворах изучаемой серии.
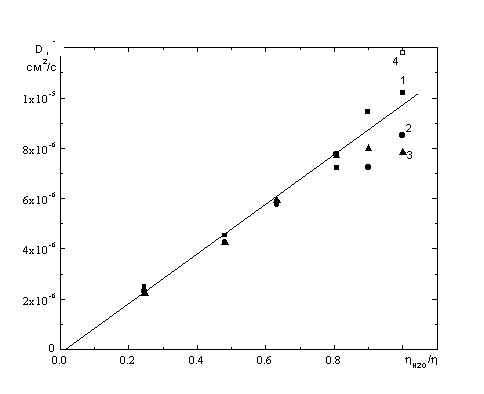 |
| Рис. 3. Коэффициенты диффузии, рассчитанные по данным рис.1. Концентрация KCl, М: 1 - 1, 2 - 0.1, 3 - 0.01. 4 – значение, приведенное в [7]. |
![]() Количественный анализ эффектов, связанных с увеличением концентрации сахарозы, проводили только для серии растворов с 1 M KCl (рис. 1а). В других сериях (рис. 1б,в) токи восстановления в минимуме полярограмм при высоких c(сах) оказались сравнимы с токами заряжения, что не обеспечивало удовлетворительной точности. Рассматривали только токи в области потенциалов -0.95 – -1.25 В, для которых пренебрегали поправкой на концентрационную поляризацию, поскольку они существенно ниже предельного диффузионного тока. Внесение обычной поправки с использованием суммарного предельного тока не обеспечивало корректности проводимого анализа, поскольку в случае параллельного восстановления реагентов разной зарядности эта процедура приводит к получению заведомо заниженных величин [31]. Соответствующая ошибка в некоторых случаях еще более чувствительна к отношению измеряемого смешанного и суммарного предельного токов, чем собственно поправка.
Количественный анализ эффектов, связанных с увеличением концентрации сахарозы, проводили только для серии растворов с 1 M KCl (рис. 1а). В других сериях (рис. 1б,в) токи восстановления в минимуме полярограмм при высоких c(сах) оказались сравнимы с токами заряжения, что не обеспечивало удовлетворительной точности. Рассматривали только токи в области потенциалов -0.95 – -1.25 В, для которых пренебрегали поправкой на концентрационную поляризацию, поскольку они существенно ниже предельного диффузионного тока. Внесение обычной поправки с использованием суммарного предельного тока не обеспечивало корректности проводимого анализа, поскольку в случае параллельного восстановления реагентов разной зарядности эта процедура приводит к получению заведомо заниженных величин [31]. Соответствующая ошибка в некоторых случаях еще более чувствительна к отношению измеряемого смешанного и суммарного предельного токов, чем собственно поправка.
![]() Выражение для плотности тока j как адиабатического, так и неадиабатического процесса можно представить уравнением (см. например [17])
Выражение для плотности тока j как адиабатического, так и неадиабатического процесса можно представить уравнением (см. например [17])
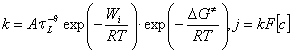 , , | (4) |
где k – наблюдаемая константа скорости; ![]() – параметр, учитывающий зависимость скорости переноса электрона от
– параметр, учитывающий зависимость скорости переноса электрона от ![]() (времени релаксации растворителя при изменении заряда сольватированной частицы [32-34]); A – независимая от свойств растворителя величина;
(времени релаксации растворителя при изменении заряда сольватированной частицы [32-34]); A – независимая от свойств растворителя величина; ![]() – работа подвода реагента к электроду;
– работа подвода реагента к электроду; ![]() – энергия активации; F – постоянная Фарадея; [с] – концентрация реагента в объеме раствора; R – универсальная газовая постоянная; T – абсолютная температура.
– энергия активации; F – постоянная Фарадея; [с] – концентрация реагента в объеме раствора; R – универсальная газовая постоянная; T – абсолютная температура.
![]() Динамический эффект растворителя количественно характеризует параметр
Динамический эффект растворителя количественно характеризует параметр ![]() ,
, ![]() для адиабатических и
для адиабатических и ![]() = 0 для неадиабатических реакций. Следовательно, отличие полученного из анализа зависимости k от
= 0 для неадиабатических реакций. Следовательно, отличие полученного из анализа зависимости k от ![]() значения
значения ![]() от нуля является признаком адиабатичности исследуемой реакции.
от нуля является признаком адиабатичности исследуемой реакции.
![]() В рамках модели Дебая
В рамках модели Дебая ![]() определяется как
определяется как
| (5) |
где es и ![]() – статическая и высокочастотная диэлектрические проницаемости среды, tD –время дебаевской (поперечной) релаксации. Для последней величины предложена формула
– статическая и высокочастотная диэлектрические проницаемости среды, tD –время дебаевской (поперечной) релаксации. Для последней величины предложена формула
| (6) |
где kB – константа Больцмана, r – радиус молекулы. Формула (6) количественно не выполняется, однако для реальных веществ наблюдается пропорциональность между ![]() и h (см., например, обсуждение в [33]). Ранее (например, в работах [13–16]) рассматривали зависимость k от
и h (см., например, обсуждение в [33]). Ранее (например, в работах [13–16]) рассматривали зависимость k от ![]() как эквивалентную зависимости k от
как эквивалентную зависимости k от ![]() , пренебрегая изменениями es и
, пренебрегая изменениями es и ![]() с ростом концентрации вязкообразующей добавки. Фактически это соответствует анализу зависимости k от
с ростом концентрации вязкообразующей добавки. Фактически это соответствует анализу зависимости k от ![]() , Ниже предпринята попытка более точного анализа.
, Ниже предпринята попытка более точного анализа.
![]() Вода, как и все протонные растворители, является недебаевской жидкостью. Ее поведение описывается в рамках представления о нескольких дебаевских временах релаксации [32–34], согласно [35,36] при 25 oC релаксация чистой воды может быть описана параметрами
Вода, как и все протонные растворители, является недебаевской жидкостью. Ее поведение описывается в рамках представления о нескольких дебаевских временах релаксации [32–34], согласно [35,36] при 25 oC релаксация чистой воды может быть описана параметрами ![]() = 78.5,
= 78.5, ![]() = 8.32·10-12 с,
= 8.32·10-12 с, ![]() = 6.18,
= 6.18, ![]() = 1.02·10-12 с,
= 1.02·10-12 с, ![]() = 4.49. Вопрос о выборе величине
= 4.49. Вопрос о выборе величине ![]() для растворителя с несколькими дебаевскими временами релаксации рассмотрен в [34,37,38]. Выбор определяется скоростью (частотой) внешнего процесса, выводящего систему из состояния равновесия, и часто диэлектрическое поведение воды, растворов неэлектролитов и не склонных к сильной ассоциации электролитов можно рассматривать как дебаевское с
для растворителя с несколькими дебаевскими временами релаксации рассмотрен в [34,37,38]. Выбор определяется скоростью (частотой) внешнего процесса, выводящего систему из состояния равновесия, и часто диэлектрическое поведение воды, растворов неэлектролитов и не склонных к сильной ассоциации электролитов можно рассматривать как дебаевское с ![]() =
= ![]() . Для анализа динамического эффекта растворителя важно, вообще говоря, не абсолютное значение
. Для анализа динамического эффекта растворителя важно, вообще говоря, не абсолютное значение ![]() , а изменение
, а изменение ![]() при изменении концентрации сахарозы.
при изменении концентрации сахарозы.
![]() Диэлектрические характеристики воды и водных растворов сахарозы и хлорида калия приводятся в справочниках [35, 37, 39]. При этом как аналог величины
Диэлектрические характеристики воды и водных растворов сахарозы и хлорида калия приводятся в справочниках [35, 37, 39]. При этом как аналог величины![]() табулируется оптическая диэлектрическая проницаемость
табулируется оптическая диэлектрическая проницаемость ![]() , определенная экспериментально по показателю преломления
, определенная экспериментально по показателю преломления ![]() для длины волны желтой линии натрия (589.3 нм) [33,34].
для длины волны желтой линии натрия (589.3 нм) [33,34].
| (7) |
![]() Используя эту величину, мы предполагаем пропорциональность
Используя эту величину, мы предполагаем пропорциональность ![]() и
и ![]() , что является общепринятым приближением.
, что является общепринятым приближением.
![]() Ниже
Ниже ![]() для исследованных растворов рассчитывали по формуле
для исследованных растворов рассчитывали по формуле
| (8) |
являющейся комбинацией формул (5) и (6). В использованном интервале концентраций веществ влияние сахарозы на величины eS и особенно ![]() выражено гораздо сильнее, чем хлорида калия, поэтому использовали значения eS и
выражено гораздо сильнее, чем хлорида калия, поэтому использовали значения eS и![]() , определенные для водных растворов сахарозы, и пренебрегали влиянием добавок хлорида калия (Табл. 1).
, определенные для водных растворов сахарозы, и пренебрегали влиянием добавок хлорида калия (Табл. 1).
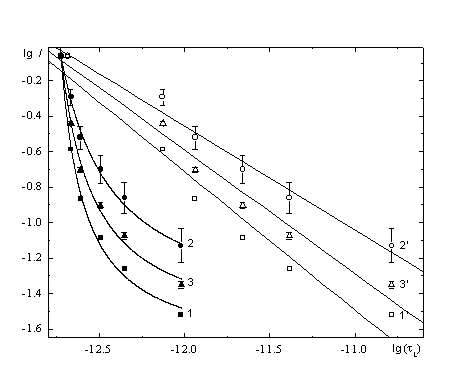
![]() Пример зависимости lgI от lg
Пример зависимости lgI от lg![]() , приведенный на рис. 4 (кривая 1), демонстрирует заведомую нелинейность. Однако это поведение не противоречит с очевидностью уравнению (4), оно может лишь указывать на зависимость составляющих энергии активации от концентрации сахарозы и(или) на отклонения динамических свойств растворителя в приэлектродном слое от объемных. Можно отметить следующие тенденции:
, приведенный на рис. 4 (кривая 1), демонстрирует заведомую нелинейность. Однако это поведение не противоречит с очевидностью уравнению (4), оно может лишь указывать на зависимость составляющих энергии активации от концентрации сахарозы и(или) на отклонения динамических свойств растворителя в приэлектродном слое от объемных. Можно отметить следующие тенденции:
![]() Высокие наблюдаемые значения
Высокие наблюдаемые значения ![]() однозначно указывают на необходимость корректировки кривой 1 рис. 4.
однозначно указывают на необходимость корректировки кривой 1 рис. 4.
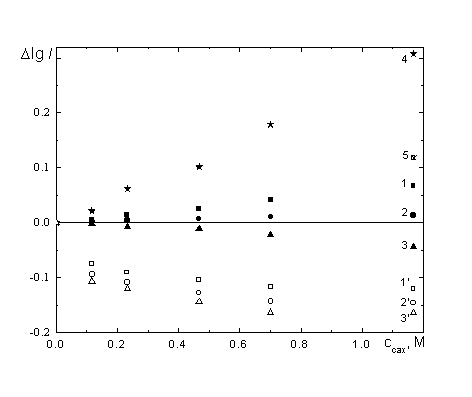 |
| Рис. 4. lg I, lg tL (1-3) и lg I, lg tL’ (1’-3’) зависимости при потенциале E = -1.1 В: (1, 1’) – без введения поправок, (2,2’) – с "монослойной" адсорбцией сахарозы, (3,3’) – с "трехслойной" адсорбцией сахарозы. |
![]() Изменение диэлектрических свойств растворителя оказывает влияние на величину энергии реорганизации. В рамках теории Маркуса [10]
Изменение диэлектрических свойств растворителя оказывает влияние на величину энергии реорганизации. В рамках теории Маркуса [10]
| (9) |
где
| (10) |
- свободная энергия реакции (разница энергии электрона в начальном и конечном состоянии), ![]() – полная энергия реорганизации системы,
– полная энергия реорганизации системы, ![]() и
и ![]() – ее внутри- и внешнесферные компоненты. Уравнение (9) применимо и для адиабатических реакций. При малых
– ее внутри- и внешнесферные компоненты. Уравнение (9) применимо и для адиабатических реакций. При малых ![]() вместо него можно использовать упрощенное уравнение
вместо него можно использовать упрощенное уравнение
| (11) |
![]() Величина
Величина ![]() равна энергии, необходимой для перехода реагента из равновесного в переходное состояние и непосредственно не связана со свойствами полярной среды. Энергию внешнесферной реорганизации растворителя
равна энергии, необходимой для перехода реагента из равновесного в переходное состояние и непосредственно не связана со свойствами полярной среды. Энергию внешнесферной реорганизации растворителя ![]() можно представить как [10,40]
можно представить как [10,40]
| (12) |
где g – величина, определяемая геометрией реагента и его расстоянием от электрода. Как видно из Таблицы 1, ![]() существенно уменьшается с ростом концентрации сахарозы.
существенно уменьшается с ростом концентрации сахарозы.
![]() Для адиабатических реакций низкозарядных реагентов при малом отклонении от равновесия можно использовать вместо уравнения (4) следующее уравнение [17]
Для адиабатических реакций низкозарядных реагентов при малом отклонении от равновесия можно использовать вместо уравнения (4) следующее уравнение [17]
| (13) |
и тем самым учесть зависимость k как от ![]() , так и
, так и ![]() . Однако для области высоких перенапряжений, отвечающих восстановлению Pt(II) на ртутном электроде (высокие
. Однако для области высоких перенапряжений, отвечающих восстановлению Pt(II) на ртутном электроде (высокие ![]() ) зависимость от
) зависимость от ![]() имеет более сложный вид и ранее в литературе не обсуждалась. В рамках теории Маркуса поправка
имеет более сложный вид и ранее в литературе не обсуждалась. В рамках теории Маркуса поправка ![]() на эффект пекаровского фактора для более высоких
на эффект пекаровского фактора для более высоких ![]() может быть представлена следующим образом:
может быть представлена следующим образом:
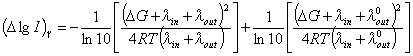 , , | (14) |
где ![]() – значение
– значение ![]() в растворе без сахарозы.
в растворе без сахарозы.
![]() Для оценки поправки использовали величины
Для оценки поправки использовали величины ![]() =
= ![]() = 80 кДж/моль, рассчитанные нами ранее для PtCl3(H2O)- [12]. Для содержащих сахарозу растворов на основании уравнения (12)
= 80 кДж/моль, рассчитанные нами ранее для PtCl3(H2O)- [12]. Для содержащих сахарозу растворов на основании уравнения (12) ![]() определяли как
определяли как ![]() , где
, где ![]() – значение
– значение ![]() в растворе без сахарозы. Различие работ приближения и отвода реагентов и продуктов Wi и Wf по нашим оценкам [12] не превышает 10 кДж/моль, т.е. невелико по сравнению с вкладом перенапряжения в свободную энергию реакции, поэтому для оценки поправки
в растворе без сахарозы. Различие работ приближения и отвода реагентов и продуктов Wi и Wf по нашим оценкам [12] не превышает 10 кДж/моль, т.е. невелико по сравнению с вкладом перенапряжения в свободную энергию реакции, поэтому для оценки поправки ![]() по формуле (14) принимали
по формуле (14) принимали ![]() . Рассчитанные в этом приближении поправки
. Рассчитанные в этом приближении поправки ![]() приведены на рис. 5, кривые 1 – 3. Они
приведены на рис. 5, кривые 1 – 3. Они ![]() плавно увеличиваются по абсолютному значению при увеличении концентрации сахарозы и малы по сравнению с наблюдаемыми изменениями тока. Отметим, что полученные с учетом высоких перенапряжений поправки гораздо меньше, чем рассчитанные по упрощенной формуле (13), справедливой исключительно при малых
плавно увеличиваются по абсолютному значению при увеличении концентрации сахарозы и малы по сравнению с наблюдаемыми изменениями тока. Отметим, что полученные с учетом высоких перенапряжений поправки гораздо меньше, чем рассчитанные по упрощенной формуле (13), справедливой исключительно при малых ![]() (кривая 4 рис. 5). Это можно обосновать соотношением
(кривая 4 рис. 5). Это можно обосновать соотношением
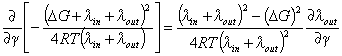 . . | (15) |
 |
| Рис. 5. Поправки |
![]() Важно также, что формально рассчитанные по формуле (14) величины
Важно также, что формально рассчитанные по формуле (14) величины ![]() меняют знак при переходе из области нормального разряда (
меняют знак при переходе из области нормального разряда (![]() ) в безактивационную область (
) в безактивационную область (![]() ), что легко видеть из формулы (15) (Подчеркнем, что зависимость пекаровского фактора от концентрации сахарозы в области нормального разряда частично компенсирует динамический эффект растворителя, что при слабом эффекте может привести к его кажущемуся отсутствию.). Однако безактивационный разряд отвечает выходу за пределы применимости теории Маркуса и, следовательно, формулы (14) и (15) становятся в этих условиях неприменимы. Поскольку эффективная энергия активации электродного процесса в безактивационной области равна нулю [41] и не зависит от
), что легко видеть из формулы (15) (Подчеркнем, что зависимость пекаровского фактора от концентрации сахарозы в области нормального разряда частично компенсирует динамический эффект растворителя, что при слабом эффекте может привести к его кажущемуся отсутствию.). Однако безактивационный разряд отвечает выходу за пределы применимости теории Маркуса и, следовательно, формулы (14) и (15) становятся в этих условиях неприменимы. Поскольку эффективная энергия активации электродного процесса в безактивационной области равна нулю [41] и не зависит от ![]() , вместо формально отрицательных значений
, вместо формально отрицательных значений ![]() (как, например, на кривой 3 рис. 5) следует использовать нулевые значения поправок.
(как, например, на кривой 3 рис. 5) следует использовать нулевые значения поправок.
![]() Работу подвода реагента к электроду
Работу подвода реагента к электроду ![]() можно в общем случае выразить как
можно в общем случае выразить как
| (16) |
где ![]() ,
, ![]() – энергия специфической адсорбции и эффективный заряд реагента [42],
– энергия специфической адсорбции и эффективный заряд реагента [42], ![]() – потенциал внешней плоскости Гельмгольца (о смысле величины эффективного заряда реагента см. в [8]. Учитывая упомянутую аналогию в адсорбционном поведении реагентов и хлорид-иона, не слишком чувствительного к соадсорбции с сахарозой, на данном этапе можно ограничиться анализом электростатической составляющей работы подвода. Ключевая величина
– потенциал внешней плоскости Гельмгольца (о смысле величины эффективного заряда реагента см. в [8]. Учитывая упомянутую аналогию в адсорбционном поведении реагентов и хлорид-иона, не слишком чувствительного к соадсорбции с сахарозой, на данном этапе можно ограничиться анализом электростатической составляющей работы подвода. Ключевая величина ![]() может изменяться из-за изменения плотности свободного заряда электрода q при адсорбции сахарозы. По теории Гуи-Чапмена, в растворе 1,1 – валентного электролита с концентрацией c
может изменяться из-за изменения плотности свободного заряда электрода q при адсорбции сахарозы. По теории Гуи-Чапмена, в растворе 1,1 – валентного электролита с концентрацией c
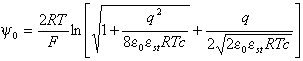 , , | (17) |
![]() При оценках использовали величины q для различных концентраций сахарозы в растворах с добавкой 1 М электролита фона по данным [21,22]. Изменение y0 при увеличении концентрации сахарозы учитывает поправка
При оценках использовали величины q для различных концентраций сахарозы в растворах с добавкой 1 М электролита фона по данным [21,22]. Изменение y0 при увеличении концентрации сахарозы учитывает поправка
| (18) |
![]() На рис. 5 (кривые 1’ – 3’) представлены значения
На рис. 5 (кривые 1’ – 3’) представлены значения ![]() , рассчитанные для
, рассчитанные для ![]() = –1. В зависимости от ориентации относительно электрода модельная величина
= –1. В зависимости от ориентации относительно электрода модельная величина ![]() , по расчетам [5], составляет от –1.1 до –0.6 для PtCl42- и от -0.6 до 0 для PtCl3(H2O)-. Для PtCl42- энергетически более выгодной является ориентация, отвечающая меньшему эффективному заряду, так что представленные величины
, по расчетам [5], составляет от –1.1 до –0.6 для PtCl42- и от -0.6 до 0 для PtCl3(H2O)-. Для PtCl42- энергетически более выгодной является ориентация, отвечающая меньшему эффективному заряду, так что представленные величины ![]() можно рассматривать как оценку сверху для любого из реагентов. Эта поправка больше, чем поправка на пекаровский фактор и противоположна ей по знаку, но также значительно меньше, чем наблюдаемое изменение тока с ростом концентрации сахарозы.
можно рассматривать как оценку сверху для любого из реагентов. Эта поправка больше, чем поправка на пекаровский фактор и противоположна ей по знаку, но также значительно меньше, чем наблюдаемое изменение тока с ростом концентрации сахарозы.
![]() В интервале использованных концентраций сахарозы ее адсорбция Г достаточно высока [21,22]. Если принять величину максимальной адсорбции равной 2.5 мкмоль/м2 [22], можно оценить заполнения поверхности сахарозой
В интервале использованных концентраций сахарозы ее адсорбция Г достаточно высока [21,22]. Если принять величину максимальной адсорбции равной 2.5 мкмоль/м2 [22], можно оценить заполнения поверхности сахарозой ![]() : при максимальной использованной концентрации они в исследуемом интервале потенциалов достигают »0.7 (Более точная оценка заполнений требует специального исследования ориентационных эффектов в слоях адсорбированной сахарозы. Здесь приведено значение, рассчитанное для предложенной в [22] ориентации).
: при максимальной использованной концентрации они в исследуемом интервале потенциалов достигают »0.7 (Более точная оценка заполнений требует специального исследования ориентационных эффектов в слоях адсорбированной сахарозы. Здесь приведено значение, рассчитанное для предложенной в [22] ориентации).
![]() По аналогии с некоторыми полифункциональными органическими молекулами [43] нельзя исключить, однако, более сложной конфигурации адсорбционного слоя сахарозы. Существенно диффузный характер распределения молекул в этом слое представляется маловероятным, поскольку на границе с воздухом сахароза демонстрирует отрицательную адсорбцию [44], а значит ее поверхностный избыток на границе ртуть-раствор стабилизирован короткодействующим хемосорбционным взаимодействием с металлом. В то же время обращает на себя внимание тот факт, что по оценкам [44] на границе с воздухом обеднен сахарозой не один монослой, а некая область, толщину которой авторы оценивают как 1 нм (это эквивалентно примерно трем монослоям сахарозы). Аналогичного распределения положительного поверхностного избытка на ртути в принципе нельзя исключить. Его причиной может быть сильное взаимодействие хемосорбированных и более удаленных от поверхности молекул сахарозы, концентрация которых в растворе достаточно велика, т.е. формирование аналога кристаллической решетки, взаимное расположение молекул в которой можно оценить по кристаллохимическим данным.
По аналогии с некоторыми полифункциональными органическими молекулами [43] нельзя исключить, однако, более сложной конфигурации адсорбционного слоя сахарозы. Существенно диффузный характер распределения молекул в этом слое представляется маловероятным, поскольку на границе с воздухом сахароза демонстрирует отрицательную адсорбцию [44], а значит ее поверхностный избыток на границе ртуть-раствор стабилизирован короткодействующим хемосорбционным взаимодействием с металлом. В то же время обращает на себя внимание тот факт, что по оценкам [44] на границе с воздухом обеднен сахарозой не один монослой, а некая область, толщину которой авторы оценивают как 1 нм (это эквивалентно примерно трем монослоям сахарозы). Аналогичного распределения положительного поверхностного избытка на ртути в принципе нельзя исключить. Его причиной может быть сильное взаимодействие хемосорбированных и более удаленных от поверхности молекул сахарозы, концентрация которых в растворе достаточно велика, т.е. формирование аналога кристаллической решетки, взаимное расположение молекул в которой можно оценить по кристаллохимическим данным.
![]() Если указанное предположение верно, то степень блокировки поверхности при фиксированном поверхностном избытке оказывается ниже, чем в случае локализации всего избытка в монослое. В предельном случае (если молекулы сахарозы располагаются в соседних слоях друг над другом) степень заполнения может снизиться втрое. Ниже эффекты блокировки оценивали для двух крайних случаев,
Если указанное предположение верно, то степень блокировки поверхности при фиксированном поверхностном избытке оказывается ниже, чем в случае локализации всего избытка в монослое. В предельном случае (если молекулы сахарозы располагаются в соседних слоях друг над другом) степень заполнения может снизиться втрое. Ниже эффекты блокировки оценивали для двух крайних случаев, ![]() = Г/ Гmax и
= Г/ Гmax и ![]() = Г/ 3Гmax.
= Г/ 3Гmax.
![]() Поскольку, как обсуждалось выше, нет оснований ожидать взаимодействия сахарозы и реагента в адсорбционном слое, можно ограничиться при анализе достаточно простым рассмотрением явления блокировки, т.е. предположить аддитивность вкладов в ток реагентов на расстояниях x1 (свободные участки поверхности) и x2 (блокированные участки) от поверхности электрода, х1 < x2 :
Поскольку, как обсуждалось выше, нет оснований ожидать взаимодействия сахарозы и реагента в адсорбционном слое, можно ограничиться при анализе достаточно простым рассмотрением явления блокировки, т.е. предположить аддитивность вкладов в ток реагентов на расстояниях x1 (свободные участки поверхности) и x2 (блокированные участки) от поверхности электрода, х1 < x2 :
| (19) |
где ![]() – профиль концентрации реагента,
– профиль концентрации реагента, ![]() ,
, ![]() – константа скорости переноса электрона для реагента на расстояниях x1 и x2 соответственно (
– константа скорости переноса электрона для реагента на расстояниях x1 и x2 соответственно (![]() >>
>> ![]() ),
), ![]() – толщина реакционного слоя.
– толщина реакционного слоя.
![]() При отсутствии эффекта адсорбции вещества скорость переноса электрона была бы равна
При отсутствии эффекта адсорбции вещества скорость переноса электрона была бы равна
| (20) |
где ![]() – профиль концентрации в отсутствие сахарозы. Поправка, учитывающая эффект адсорбции, равна
– профиль концентрации в отсутствие сахарозы. Поправка, учитывающая эффект адсорбции, равна
 | (21) |
или
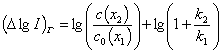 . . | (22) |
![]() Первое слагаемое в уравнении (22) описывает конкурентную адсорбцию реагента и сахарозы, второе – замедленный перенос через пленку адсорбата. Отличие между
Первое слагаемое в уравнении (22) описывает конкурентную адсорбцию реагента и сахарозы, второе – замедленный перенос через пленку адсорбата. Отличие между ![]() и
и ![]() будет достаточно резким в первую очередь из-за зависимости трансмиссионного коэффициента от расстояния:
будет достаточно резким в первую очередь из-за зависимости трансмиссионного коэффициента от расстояния:
| (23) |
где d – характерное расстояние, на котором трансмиссионный коэффициент уменьшается в e раз. Для аквахлоридных комплексов Pt(II) d составляет около 0.05 нм [45], величина ![]() по крайней мере не меньше толщины слоя адсорбированных в планарной ориентации молекул сахарозы (0.3 нм). Это дает оценку
по крайней мере не меньше толщины слоя адсорбированных в планарной ориентации молекул сахарозы (0.3 нм). Это дает оценку ![]() , что означает пренебрежимо малый вклад частиц реагента, расположенных на дальнем расстоянии от электрода, которым можно пренебречь. Еще более сильного торможения с увеличением х следует ожидать с учетом проигрыша в работе подвода (на расстоянии х2 мала или равна нулю энергия адсорбции реагента), тем более в упомянутом выше случае локализации поверхностного избытка вне пределов одного монослоя.
, что означает пренебрежимо малый вклад частиц реагента, расположенных на дальнем расстоянии от электрода, которым можно пренебречь. Еще более сильного торможения с увеличением х следует ожидать с учетом проигрыша в работе подвода (на расстоянии х2 мала или равна нулю энергия адсорбции реагента), тем более в упомянутом выше случае локализации поверхностного избытка вне пределов одного монослоя.
![]() С учетом слабого взаимодействия сахарозы и реагента в адсорбированном состоянии величину
С учетом слабого взаимодействия сахарозы и реагента в адсорбированном состоянии величину 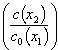 можно принять равной доле свободной поверхности электрода, т.е. использовать простую формулу
можно принять равной доле свободной поверхности электрода, т.е. использовать простую формулу
| (24) |
![]() Рассчитанные по формуле (24) для
Рассчитанные по формуле (24) для ![]() = 2.5 мкмоль/м2 из данных [22] "блокировочные" поправки в комбинации с меньшими по величине поправками (рис. 4) не позволяют линеаризовать зависимости lg I от lg
= 2.5 мкмоль/м2 из данных [22] "блокировочные" поправки в комбинации с меньшими по величине поправками (рис. 4) не позволяют линеаризовать зависимости lg I от lg ![]() (кривые 2 и 3 рис. 4). Наклоны кривых на всех участках в результате внесения поправок существенно снижаются, но для всех потенциалов при не слишком высоких концентраций сахарозы по-прежнему отвечают
(кривые 2 и 3 рис. 4). Наклоны кривых на всех участках в результате внесения поправок существенно снижаются, но для всех потенциалов при не слишком высоких концентраций сахарозы по-прежнему отвечают ![]() > 1. Таким образом, очевидные отклонения от уравнения (4) сохраняются, хотя зависимость различных составляющих энергии активации и изменения доли свободной поверхности поправками учтены.
> 1. Таким образом, очевидные отклонения от уравнения (4) сохраняются, хотя зависимость различных составляющих энергии активации и изменения доли свободной поверхности поправками учтены.
![]() Отмеченная выше тенденция для зависимости наклона lg I, lg
Отмеченная выше тенденция для зависимости наклона lg I, lg ![]() -кривых от потенциала при фиксированном
-кривых от потенциала при фиксированном ![]() после внесения поправок сохраняется.
после внесения поправок сохраняется.
![]() Очевидным внутренним противоречием проведенного выше анализа является использование величин объемной вязкости для расчета времен релаксации в реакционном слое, обогащенном сахарозой. Очевидно, что в непосредственной близости от реагирующей частицы, локализованной в ближайшем к поверхности монослое, релаксация растворителя происходит медленнее, чем в объеме, т.е. с некоторой долей условности можно говорить о повышенной "локальной вязкости" в реакционном слое.
Очевидным внутренним противоречием проведенного выше анализа является использование величин объемной вязкости для расчета времен релаксации в реакционном слое, обогащенном сахарозой. Очевидно, что в непосредственной близости от реагирующей частицы, локализованной в ближайшем к поверхности монослое, релаксация растворителя происходит медленнее, чем в объеме, т.е. с некоторой долей условности можно говорить о повышенной "локальной вязкости" в реакционном слое.
![]() Оценка эффекта "локальной вязкости" может быть на сегодняшний день дана лишь в очень грубом приближении, имеющем, очевидно, смысл, только для немонослойного распределения поверхностного избытка сахарозы (в противном случае применимость макроскопического понятия вязкости для расчета времен релаксации особенно сомнительна). Целью проводимых ниже оценок является выяснение принципиального вопроса о том, позволяет ли такой не вполне количественный учет локальной вязкости получить зависимость, более близкую к предсказываемой уравнением (4), чем обработка экспериментальных данных с использованием объемных значений вязкости.
Оценка эффекта "локальной вязкости" может быть на сегодняшний день дана лишь в очень грубом приближении, имеющем, очевидно, смысл, только для немонослойного распределения поверхностного избытка сахарозы (в противном случае применимость макроскопического понятия вязкости для расчета времен релаксации особенно сомнительна). Целью проводимых ниже оценок является выяснение принципиального вопроса о том, позволяет ли такой не вполне количественный учет локальной вязкости получить зависимость, более близкую к предсказываемой уравнением (4), чем обработка экспериментальных данных с использованием объемных значений вязкости.
![]() При оценке полагали, что концентрация сахарозы в реакционном слое повышена по сравнению с объемной концентрацией на величину, определяемую поверхностным избытком и объемом обогащенного слоя (толщину последнего полагали, с учетом вышесказанного, равной 1 нм). Все существенные для оценки параметры определяли для полученных исправленных концентраций таким же образом, как описано выше. Соответствующие параметры (обозначенные штрихом), оцененные для потенциала -0.95 В представлены в Таблице 2.
При оценке полагали, что концентрация сахарозы в реакционном слое повышена по сравнению с объемной концентрацией на величину, определяемую поверхностным избытком и объемом обогащенного слоя (толщину последнего полагали, с учетом вышесказанного, равной 1 нм). Все существенные для оценки параметры определяли для полученных исправленных концентраций таким же образом, как описано выше. Соответствующие параметры (обозначенные штрихом), оцененные для потенциала -0.95 В представлены в Таблице 2.
Таблица 2. Оценки эффективных диэлектрических и релаксационных свойств приповерхностных слоев растворов сахарозы в предположении о повышенной "локальной вязкости"
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| * Наименее точны оценки для раствора самой высокой концентрации, так как при выбранной толщине обогащенного слоя с’(сах) близко к растворимости сахарозы. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
![]() Кривые 1’–3’ на рис. 4 построены с внесением таких же поправок в величины тока, как и для кривых 1–3, но с использованием
Кривые 1’–3’ на рис. 4 построены с внесением таких же поправок в величины тока, как и для кривых 1–3, но с использованием ![]() =
= ![]() '. Форма кривых 1’ – 3’ оказывается значительно ближе к линейной, а их наклоны для всего обсуждаемого интервала отвечают
'. Форма кривых 1’ – 3’ оказывается значительно ближе к линейной, а их наклоны для всего обсуждаемого интервала отвечают ![]() < 1, хотя точность определения значений наклонов не слишком высока из-за неоднозначности поправок на блокировку.
< 1, хотя точность определения значений наклонов не слишком высока из-за неоднозначности поправок на блокировку.
![]() Зависимости
Зависимости ![]() , рассчитанных из кривых 1’ – 3’, от потенциала представлены на рис. 6. Очевидно, что вне зависимости от методики внесения поправок форма полученной зависимости свидетельствует о существенном изменении степени адиабатичности процесса с потенциалом. Как отмечалось выше, максимальным
, рассчитанных из кривых 1’ – 3’, от потенциала представлены на рис. 6. Очевидно, что вне зависимости от методики внесения поправок форма полученной зависимости свидетельствует о существенном изменении степени адиабатичности процесса с потенциалом. Как отмечалось выше, максимальным ![]() отвечает область потенциалов, предшествующих десорбции доминирующего реагента, а при высоких отрицательных потенциалах
отвечает область потенциалов, предшествующих десорбции доминирующего реагента, а при высоких отрицательных потенциалах ![]() значимо снижается. Этот важный качественный результат устойчив к внесению поправок, но может рассматриваться как полуколичественный только при учете эффектов "локальной вязкости".
значимо снижается. Этот важный качественный результат устойчив к внесению поправок, но может рассматриваться как полуколичественный только при учете эффектов "локальной вязкости".
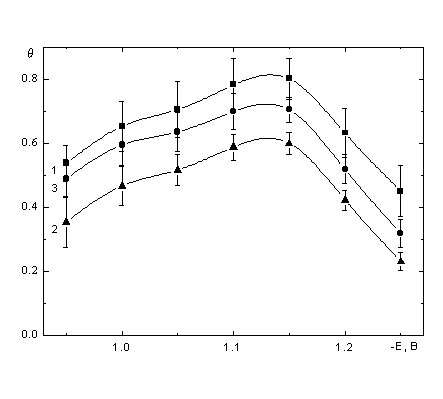 |
| Рис. 6. Cредние величины q, определенных из lg I, lg tL’ зависимостей при разных потенциалах до введения поправок (1), и после введения поправки на "монослойную" (2) и "трехслойную" адсорбцию сахарозы. |
![]() Необходимо специально оговорить возможность включения точки для чистой воды в серию зависимости от вязкости. В работе [14] такая точка на приведенных графиках отсутствует, а точка для раствора самой низкой по сахарозе концентрации явно выпадает из общей зависимости. Действительно, статистическая достоверность любых построений с учетом этой точки крайне сомнительна, т.к. при переходе к растворам содержащим сахарозу, степень блокировки поверхности изменяется очень резко, внося вклад, сравнимый с влиянием вязкости на величину тока на свободной поверхности. Однако, как видно из кривых 1’–3’ рис. 6, при учете возможности изменения локальной вязкости отклонения точки для чистого раствора оказываются не столь велики. На наш взгляд, это указывает на разумность внесения приближенной поправки на локальную вязкость предложенным выше способом.
Необходимо специально оговорить возможность включения точки для чистой воды в серию зависимости от вязкости. В работе [14] такая точка на приведенных графиках отсутствует, а точка для раствора самой низкой по сахарозе концентрации явно выпадает из общей зависимости. Действительно, статистическая достоверность любых построений с учетом этой точки крайне сомнительна, т.к. при переходе к растворам содержащим сахарозу, степень блокировки поверхности изменяется очень резко, внося вклад, сравнимый с влиянием вязкости на величину тока на свободной поверхности. Однако, как видно из кривых 1’–3’ рис. 6, при учете возможности изменения локальной вязкости отклонения точки для чистого раствора оказываются не столь велики. На наш взгляд, это указывает на разумность внесения приближенной поправки на локальную вязкость предложенным выше способом.
![]() Строго говоря, самосогласованный учет "локальной вязкости" требует модифицировать также и поправку на изменение пекаровского фактора. Проведенные оценки показали, что модифицированная таким образом поправка крайне мала и заведомо лежит в пределах неопределенности, вносимой поправкой на блокировку, поэтому на данном этапе такое усложнение расчета не имеет смысла.
Строго говоря, самосогласованный учет "локальной вязкости" требует модифицировать также и поправку на изменение пекаровского фактора. Проведенные оценки показали, что модифицированная таким образом поправка крайне мала и заведомо лежит в пределах неопределенности, вносимой поправкой на блокировку, поэтому на данном этапе такое усложнение расчета не имеет смысла.
![]() На основании проведенного анализа можно с уверенностью утверждать, что значения
На основании проведенного анализа можно с уверенностью утверждать, что значения ![]() во всей области потенциалов минимума тока заведомо не равны нулю. Таким образом, экспериментально установлен выраженный динамический эффект растворителя в системе, сильно отличающейся по свойствам от исследованных ранее другими авторами. Для реакции при высоких перенапряжениях такое исследование предпринято впервые.
во всей области потенциалов минимума тока заведомо не равны нулю. Таким образом, экспериментально установлен выраженный динамический эффект растворителя в системе, сильно отличающейся по свойствам от исследованных ранее другими авторами. Для реакции при высоких перенапряжениях такое исследование предпринято впервые.
![]() Как отмечалось выше, проанализировать зависимость тока от вязкости для растворов с более низкой концентрацией КCl не представляется возможным. Следует, однако, сделать некоторые качественные заключения. Данные рис. 1б (0,1 М KCl) указывают на практически полную независимость тока от концентрации сахарозы при потенциалах -1.4 – -1.5 В, что отвечает
Как отмечалось выше, проанализировать зависимость тока от вязкости для растворов с более низкой концентрацией КCl не представляется возможным. Следует, однако, сделать некоторые качественные заключения. Данные рис. 1б (0,1 М KCl) указывают на практически полную независимость тока от концентрации сахарозы при потенциалах -1.4 – -1.5 В, что отвечает ![]() =0. Для рассмотренной выше серии с 1 M KCl (рис. 1а) в этой области происходит выход на предельный диффузионный ток, поэтому проследить кинетику процесса не удается. Можно, однако, ожидать, что кривые на рис. 6 при еще более отрицательных потенциалах приблизятся к нулевому значению
=0. Для рассмотренной выше серии с 1 M KCl (рис. 1а) в этой области происходит выход на предельный диффузионный ток, поэтому проследить кинетику процесса не удается. Можно, однако, ожидать, что кривые на рис. 6 при еще более отрицательных потенциалах приблизятся к нулевому значению ![]() .
.
![]() Ранее нами было проанализировано строение реакционного слоя для процесса электровосстановления ансамбля аквахлоридных комплексов Pt(II) [42] в водных растворах. Результаты анализа позволили сделать вывод о том, что закономерности исследуемого процесса во многом определяются зависимой от потенциала электрода адсорбцией комплексов Pt(II). Однако попытки моделирования реакции восстановления в рамках представлений о неадиабатических процессах не дали удовлетворительных результатов. Предварительные расчеты электронного перекрывания указывают на то, что в адсорбированном состоянии реагента происходит адиабатический перенос электрона, и полученные в настоящей работе результаты экспериментально обосновывают это заключение, что позволяет более определенно выбрать сценарий моделирования процесса с рассмотрением адиабатического переноса электрона.
Ранее нами было проанализировано строение реакционного слоя для процесса электровосстановления ансамбля аквахлоридных комплексов Pt(II) [42] в водных растворах. Результаты анализа позволили сделать вывод о том, что закономерности исследуемого процесса во многом определяются зависимой от потенциала электрода адсорбцией комплексов Pt(II). Однако попытки моделирования реакции восстановления в рамках представлений о неадиабатических процессах не дали удовлетворительных результатов. Предварительные расчеты электронного перекрывания указывают на то, что в адсорбированном состоянии реагента происходит адиабатический перенос электрона, и полученные в настоящей работе результаты экспериментально обосновывают это заключение, что позволяет более определенно выбрать сценарий моделирования процесса с рассмотрением адиабатического переноса электрона.
![]() Выполненный выше анализ имеет самостоятельное методическое значение, а предложенные новые подходы к внесению разного рода поправок могут быть использованы для более детального анализа опубликованных ранее данных по зависимости скоростей электродных процессов от вязкости. В частности, значительные поправки на блокировку поверхности ртути сахарозой и учет локальной вязкости позволят, по нашим предварительным расчетом, извлечь существенную новую информацию о восстановлении комплекса Cr(III)EDTA- путем обработки данных работы [14].
Выполненный выше анализ имеет самостоятельное методическое значение, а предложенные новые подходы к внесению разного рода поправок могут быть использованы для более детального анализа опубликованных ранее данных по зависимости скоростей электродных процессов от вязкости. В частности, значительные поправки на блокировку поверхности ртути сахарозой и учет локальной вязкости позволят, по нашим предварительным расчетом, извлечь существенную новую информацию о восстановлении комплекса Cr(III)EDTA- путем обработки данных работы [14].
![]() При проведении новых экспериментов в растворах сахарозы следует также учесть возможность некоторого упрощения их постановки. Очевидно, что все возможные эффекты, связанные с введением сахарозы, при больших концентрациях оказываются близки к насыщению (эффект блокировки и изменения электростатического взаимодействия) или, по крайней мере, остаются малыми (эффект пекаровского фактора). При высоких концентрациях значительно меньше оказывается и возможное изменение локальной вязкости, так как поверхностный избыток вязкообразователя увеличивается с концентрацией медленно. Поэтому в принципе грубые оценки степени адиабатичности могут быть выполнены по данным для нескольких растворов достаточно высоких концентраций без детализации всех рассмотренных выше поправок.
При проведении новых экспериментов в растворах сахарозы следует также учесть возможность некоторого упрощения их постановки. Очевидно, что все возможные эффекты, связанные с введением сахарозы, при больших концентрациях оказываются близки к насыщению (эффект блокировки и изменения электростатического взаимодействия) или, по крайней мере, остаются малыми (эффект пекаровского фактора). При высоких концентрациях значительно меньше оказывается и возможное изменение локальной вязкости, так как поверхностный избыток вязкообразователя увеличивается с концентрацией медленно. Поэтому в принципе грубые оценки степени адиабатичности могут быть выполнены по данным для нескольких растворов достаточно высоких концентраций без детализации всех рассмотренных выше поправок.
![]() Работа выполнена при поддержке гранта РФФИ 02–03–33284. Авторы благодарны проф. Дамаскина Б. Б. за ценное обсуждение.
Работа выполнена при поддержке гранта РФФИ 02–03–33284. Авторы благодарны проф. Дамаскина Б. Б. за ценное обсуждение.