![]() Установлено, что в метастабильных долгоживущих истинных растворах изополивольфраматов при рН ~ 0,5 на платиновом электроде в области адсорбции водорода реализуются два типа редокс-процессов – с образованием растворенных восстановленных форм вольфраматов и с формированием твердых продуктов. Последние накапливаются на поверхности платины в виде кристаллических пленок, способных к обратимому перезаряжению. Дана общая характеристика этих пленок методами циклической вольтамперометрии, рентгеновской дифрактометрии и сканирующей туннельной микроскопии. На основании спектрофотометрического анализа растворов наблюдаемые в водородной области потенциалов обратимые редокс-превращения отнесены к присутствующим в системе растворенным гепта- и декавольфраматам.
Установлено, что в метастабильных долгоживущих истинных растворах изополивольфраматов при рН ~ 0,5 на платиновом электроде в области адсорбции водорода реализуются два типа редокс-процессов – с образованием растворенных восстановленных форм вольфраматов и с формированием твердых продуктов. Последние накапливаются на поверхности платины в виде кристаллических пленок, способных к обратимому перезаряжению. Дана общая характеристика этих пленок методами циклической вольтамперометрии, рентгеновской дифрактометрии и сканирующей туннельной микроскопии. На основании спектрофотометрического анализа растворов наблюдаемые в водородной области потенциалов обратимые редокс-превращения отнесены к присутствующим в системе растворенным гепта- и декавольфраматам.
![]() Ключевые слова: платиновый электрод, изополивольфраматы, нестехиометрический оксид вольфрама.
Ключевые слова: платиновый электрод, изополивольфраматы, нестехиометрический оксид вольфрама.
![]() В последние годы большое внимание привлекают каталитические системы на основе полиоксометаллатов [1], в том числе модифицированные ими электроды [2, 3]. Иммобилизация гетерополианионов (ГПА) на твердых поверхностях и электрокаталитические свойства образующихся при этом систем относительно хорошо изучены. Однако до сих пор не проводились сравнительные исследования электрохимического поведения различных изополианионов (ИПА) в кислых средах, возможно вследствие того, что в этих системах реализуются более сложные равновесия и имеются существенные ограничения по растворимости [4 – 8]. В литературе также практически отсутствуют сведения о поведении поливольфраматов на платиновом электроде, который является удобной модельной системой для исследования адсорбции благодаря наличию характерных областей адсорбции-десорбции водорода и кислорода, чувствительных к присутствию посторонних частиц на поверхности.
В последние годы большое внимание привлекают каталитические системы на основе полиоксометаллатов [1], в том числе модифицированные ими электроды [2, 3]. Иммобилизация гетерополианионов (ГПА) на твердых поверхностях и электрокаталитические свойства образующихся при этом систем относительно хорошо изучены. Однако до сих пор не проводились сравнительные исследования электрохимического поведения различных изополианионов (ИПА) в кислых средах, возможно вследствие того, что в этих системах реализуются более сложные равновесия и имеются существенные ограничения по растворимости [4 – 8]. В литературе также практически отсутствуют сведения о поведении поливольфраматов на платиновом электроде, который является удобной модельной системой для исследования адсорбции благодаря наличию характерных областей адсорбции-десорбции водорода и кислорода, чувствительных к присутствию посторонних частиц на поверхности.
![]() В настоящей работе исследовано поведение гладкого платинового электрода в истинных растворах изополивольфраматов в 0,5 М Н2SO4 при рН ~ 0,5, а также дана характеристика поверхности Pt, модифицированной изополивольфраматами. Это оказалось возможным благодаря разработке методики приготовления долгоживущих кислых метастабильных растворов ИПА, не содержащих коллоидной вольфрамовой кислоты (В условиях равновесия в таких растворах происходит образование малорастворимой вольфрамовой кислоты (ее состав иногда представляют формулой Н2WO4, в действительности, по-видимому, образуются гидратированные оксиды WO3*nH2O).). Платина, соосажденная с твердыми оксовольфраматными соединениями из коллоидных растворов вольфрамовой кислоты, демонстрирует необычные и привлекательные электрокаталитические свойства [9, 10]. Представляет интерес выявление закономерностей адсорбционного поведения таких систем, приготовленных в более контролируемых условиях из истинных растворов.
В настоящей работе исследовано поведение гладкого платинового электрода в истинных растворах изополивольфраматов в 0,5 М Н2SO4 при рН ~ 0,5, а также дана характеристика поверхности Pt, модифицированной изополивольфраматами. Это оказалось возможным благодаря разработке методики приготовления долгоживущих кислых метастабильных растворов ИПА, не содержащих коллоидной вольфрамовой кислоты (В условиях равновесия в таких растворах происходит образование малорастворимой вольфрамовой кислоты (ее состав иногда представляют формулой Н2WO4, в действительности, по-видимому, образуются гидратированные оксиды WO3*nH2O).). Платина, соосажденная с твердыми оксовольфраматными соединениями из коллоидных растворов вольфрамовой кислоты, демонстрирует необычные и привлекательные электрокаталитические свойства [9, 10]. Представляет интерес выявление закономерностей адсорбционного поведения таких систем, приготовленных в более контролируемых условиях из истинных растворов.
![]() Согласно литературным данным [11 – 13], для различных ИПА окислительно-восстановительный потенциал первого редокс-перехода WVI/WV в концентрированных солянокислых растворах (рН
Согласно литературным данным [11 – 13], для различных ИПА окислительно-восстановительный потенциал первого редокс-перехода WVI/WV в концентрированных солянокислых растворах (рН ![]() 0) отрицательнее обратимого водородного. По данным [14] восстановление паравольфрамата Nа10[(H2)W12O42] в ацетатных буферных растворах при рН от 4 до 6 протекает при высоких отрицательных потенциалах, предшествующих выделению водорода на стеклоуглероде (по крайней мере на 0,3 – 0,5 В более отрицательных, чем равновесный потенциал системы H3O+/H2 при соответствующих рН). Даже если предположить существенную рН-зависимость равновесного потенциала WVI/WV (> 100 мВ/рН), на основании этих данных можно было бы исключить электродные превращения ИПА в области адсорбции водорода и кислорода. Однако состояние комплексных ионов в концентрированных солянокислых растворах [11 – 13] может отличаться от известного строения ИПА. Не вполне однозначна и интерпретация данных [14], поскольку восстановление паравольфрамата при рН 4 – 6 характеризуется достаточно сложными кинетическими закономерностями, и формальные потенциалы могут быть смещены относительно равновесных. В настоящей работе предпринята попытка оценить соответствующий потенциал для сернокислых растворов, в которых традиционно проводятся электрокаталитические исследования.
0) отрицательнее обратимого водородного. По данным [14] восстановление паравольфрамата Nа10[(H2)W12O42] в ацетатных буферных растворах при рН от 4 до 6 протекает при высоких отрицательных потенциалах, предшествующих выделению водорода на стеклоуглероде (по крайней мере на 0,3 – 0,5 В более отрицательных, чем равновесный потенциал системы H3O+/H2 при соответствующих рН). Даже если предположить существенную рН-зависимость равновесного потенциала WVI/WV (> 100 мВ/рН), на основании этих данных можно было бы исключить электродные превращения ИПА в области адсорбции водорода и кислорода. Однако состояние комплексных ионов в концентрированных солянокислых растворах [11 – 13] может отличаться от известного строения ИПА. Не вполне однозначна и интерпретация данных [14], поскольку восстановление паравольфрамата при рН 4 – 6 характеризуется достаточно сложными кинетическими закономерностями, и формальные потенциалы могут быть смещены относительно равновесных. В настоящей работе предпринята попытка оценить соответствующий потенциал для сернокислых растворов, в которых традиционно проводятся электрокаталитические исследования.
![]() В работе использовали гладкий платиновый электрод из фольги с видимой поверхностью 8,5 см2 и фактором шероховатости ~ 2,1 – 2,3, определенным по десорбции водорода. Вспомогательным электродом служила платиновая проволока, а электродом сравнения – обратимый водородный электрод в растворе фона 0,5 М H2SO4 (измеренные относительно этого электрода потенциалы обозначены Er). Для очистки поверхности Pt перед каждой серией опытов электрод подвергали травлению в кипящей царской водке в течение 30 – 40 с. Затем электрод промывали и поляризовали попеременно анодно и катодно при плотности тока 0,04 мА/см2 видимой поверхности в растворе 0,05 М H2SO4 по 10 минут, с промежуточной заменой раствора. При последовательных измерениях в растворах с разной концентрацией перед каждым новым опытом электрод помещали в раствор 0,01 M NaOH на 1 – 2 минуты, чтобы разрушить образующуюся на поверхности пленку вольфрамата, а затем поляризовали анодно и катодно. Достигнутую чистоту поверхности электрода контролировали по потенциодинамической кривой в растворе фона.
В работе использовали гладкий платиновый электрод из фольги с видимой поверхностью 8,5 см2 и фактором шероховатости ~ 2,1 – 2,3, определенным по десорбции водорода. Вспомогательным электродом служила платиновая проволока, а электродом сравнения – обратимый водородный электрод в растворе фона 0,5 М H2SO4 (измеренные относительно этого электрода потенциалы обозначены Er). Для очистки поверхности Pt перед каждой серией опытов электрод подвергали травлению в кипящей царской водке в течение 30 – 40 с. Затем электрод промывали и поляризовали попеременно анодно и катодно при плотности тока 0,04 мА/см2 видимой поверхности в растворе 0,05 М H2SO4 по 10 минут, с промежуточной заменой раствора. При последовательных измерениях в растворах с разной концентрацией перед каждым новым опытом электрод помещали в раствор 0,01 M NaOH на 1 – 2 минуты, чтобы разрушить образующуюся на поверхности пленку вольфрамата, а затем поляризовали анодно и катодно. Достигнутую чистоту поверхности электрода контролировали по потенциодинамической кривой в растворе фона.
![]() Потенциодинамические кривые измеряли при скоростях развертки 20 – 100 мВ/с в интервале Er от 0,05 до 1,3 В на потенциостате PAR273EG&G и записывали с помощью двухкоординатного самописца.
Потенциодинамические кривые измеряли при скоростях развертки 20 – 100 мВ/с в интервале Er от 0,05 до 1,3 В на потенциостате PAR273EG&G и записывали с помощью двухкоординатного самописца.
![]() Были проведены две серии экспериментов. В первой исследовались растворы солей вольфраматов Na2WO4 (W) и (NH4)10[(H2)W12O42] (W12) различной концентрации (0,1
Были проведены две серии экспериментов. В первой исследовались растворы солей вольфраматов Na2WO4 (W) и (NH4)10[(H2)W12O42] (W12) различной концентрации (0,1 ![]() 1мМ) в 0,5 М H2SO4 (Ниже для сокращенного обозначения различных полимерных форм оксовольфраматов используется традиционная запись Wn, где n – число атомов вольфрама в изополианионе.). Отметим, что для удобства сравнения результатов для Na2WO4 "эквивалентную" концентрацию рассчитывали на двенадцать атомов вольфрама, поэтому в дальнейшем будем обозначать ее как Мэ. Во второй серии опытов с отмывкой электрода исследовались 1 мМ растворы указанных солей в 0,5 М H2SO4. Соблюдали следующую последовательность операций. После предварительной анодно-катодной поляризации и деаэрирования раствора аргоном измеряли потенциодинамическую кривую в фоне, затем заменяли фоновый раствор на раствор поливольфрамата, деаэрировали в течение 20 – 30 минут и измеряли потенциодинамические кривые при разных скоростях развертки и различных временах задержки при потенциале 0,05 В (0 и 60 с). Цепь после завершения циклирования размыкали при Er = 0,05 В, ячейку продували аргоном. При разомкнутой цепи раствор поливольфрамата сливали, промывали электрод деаэрированным фоновым раствором, затем заполняли им ячейку и проводили такую же серию измерений, что и в растворе вольфрамата. При такой постановке эксперимента нельзя было исключить смещение потенциала электрода в ходе отмывки в сторону более положительных значений, что могло привести к изменению состояния адсорбционного слоя (частичная десорбция). Однако поскольку опыты с отмывкой были направлены на получение общей характеристики прочно хемосорбированных частиц и полислойных пленок, смещение потенциала, видимо, не играло принципиальной роли.
1мМ) в 0,5 М H2SO4 (Ниже для сокращенного обозначения различных полимерных форм оксовольфраматов используется традиционная запись Wn, где n – число атомов вольфрама в изополианионе.). Отметим, что для удобства сравнения результатов для Na2WO4 "эквивалентную" концентрацию рассчитывали на двенадцать атомов вольфрама, поэтому в дальнейшем будем обозначать ее как Мэ. Во второй серии опытов с отмывкой электрода исследовались 1 мМ растворы указанных солей в 0,5 М H2SO4. Соблюдали следующую последовательность операций. После предварительной анодно-катодной поляризации и деаэрирования раствора аргоном измеряли потенциодинамическую кривую в фоне, затем заменяли фоновый раствор на раствор поливольфрамата, деаэрировали в течение 20 – 30 минут и измеряли потенциодинамические кривые при разных скоростях развертки и различных временах задержки при потенциале 0,05 В (0 и 60 с). Цепь после завершения циклирования размыкали при Er = 0,05 В, ячейку продували аргоном. При разомкнутой цепи раствор поливольфрамата сливали, промывали электрод деаэрированным фоновым раствором, затем заполняли им ячейку и проводили такую же серию измерений, что и в растворе вольфрамата. При такой постановке эксперимента нельзя было исключить смещение потенциала электрода в ходе отмывки в сторону более положительных значений, что могло привести к изменению состояния адсорбционного слоя (частичная десорбция). Однако поскольку опыты с отмывкой были направлены на получение общей характеристики прочно хемосорбированных частиц и полислойных пленок, смещение потенциала, видимо, не играло принципиальной роли.
![]() Все растворы готовили на бидистиллированной воде, дополнительно очищенной на установке Milli-Q. Для приготовления растворов серной кислоты использовали 18 M H2SO4, дважды перегнанную в вакууме. Использовали (NH4)10[(H2)W12O42] *n H2O марки ч.д.а. и Na2WO4*2 H2O (PARC, p.a.).
Все растворы готовили на бидистиллированной воде, дополнительно очищенной на установке Milli-Q. Для приготовления растворов серной кислоты использовали 18 M H2SO4, дважды перегнанную в вакууме. Использовали (NH4)10[(H2)W12O42] *n H2O марки ч.д.а. и Na2WO4*2 H2O (PARC, p.a.).
![]() "Долгоживущие" кислые растворы изополивольфраматов в 0,5 М H2SO4 готовили по следующей методике. В колбу помещали навеску вольфрамата, добавляли воду (около 80% конечного объема раствора), подогревали колбу в пламени горелки почти до кипения, при этом наблюдалось полное растворение вещества. Затем в горячий раствор вольфрамата быстро добавляли необходимое количество 18 М H2SO4, доводили до метки водой, перемешивали. Быстрое приливание кислоты продлевает время жизни растворов, поскольку приводит к образованию декамерного вольфрамата W10 (схема 1), который нестабилен и разлагается на оксовольфраматные фрагменты с меньшей степенью полимеризации [4]. При старении раствора равновесие постепенно смещается в сторону образования нерастворимого продукта.
"Долгоживущие" кислые растворы изополивольфраматов в 0,5 М H2SO4 готовили по следующей методике. В колбу помещали навеску вольфрамата, добавляли воду (около 80% конечного объема раствора), подогревали колбу в пламени горелки почти до кипения, при этом наблюдалось полное растворение вещества. Затем в горячий раствор вольфрамата быстро добавляли необходимое количество 18 М H2SO4, доводили до метки водой, перемешивали. Быстрое приливание кислоты продлевает время жизни растворов, поскольку приводит к образованию декамерного вольфрамата W10 (схема 1), который нестабилен и разлагается на оксовольфраматные фрагменты с меньшей степенью полимеризации [4]. При старении раствора равновесие постепенно смещается в сторону образования нерастворимого продукта.
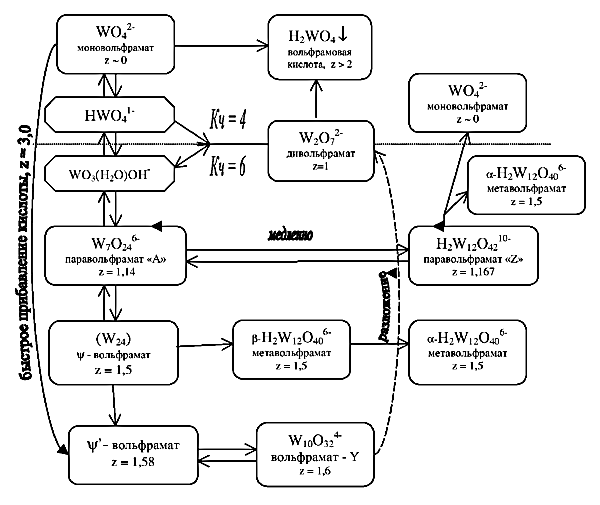 |
| Схема 1. Взаимопревращения изополивольфраматов в подкисленном водном растворе. |
![]() Состав растворов изополивольфраматов и начало образования коллоида Н2WO4 контролировали спектрофотометрически. Для регистрации спектров в ультрафиолетовой области использовали спектрофотометр Unicam SP 1800 Ultraviolet Spectrophotometer, который позволял измерять оптическую плотность в интервале длин волн от 200 до 800 нм. Толщина кварцевых кювет была равна 1,0 см. Спектры мутности в видимой области измеряли на спектрофотоколориметре КФК-3.
Состав растворов изополивольфраматов и начало образования коллоида Н2WO4 контролировали спектрофотометрически. Для регистрации спектров в ультрафиолетовой области использовали спектрофотометр Unicam SP 1800 Ultraviolet Spectrophotometer, который позволял измерять оптическую плотность в интервале длин волн от 200 до 800 нм. Толщина кварцевых кювет была равна 1,0 см. Спектры мутности в видимой области измеряли на спектрофотоколориметре КФК-3.
![]() Для выяснения природы пленки, которая образуется на поверхности платины в растворах изополивольфраматов, извлеченные модифицированные электроды исследовали методами рентгеновской дифракции и сканирующей туннельной микроскопии ex situ. Рентгеновские дифрактограммы регистрировали на дифрактометре ДРОН-2 (СuKa - излучение) в интервале углов 15 < 2q < 90. Расшифровку дифрактограмм проводили с использованием электронной картотеки PCPDF – 95 и базы данных JCSD [15]. СТМ-изображения поверхности регистрировали на микроскопе LitScan2 [16].
Для выяснения природы пленки, которая образуется на поверхности платины в растворах изополивольфраматов, извлеченные модифицированные электроды исследовали методами рентгеновской дифракции и сканирующей туннельной микроскопии ex situ. Рентгеновские дифрактограммы регистрировали на дифрактометре ДРОН-2 (СuKa - излучение) в интервале углов 15 < 2q < 90. Расшифровку дифрактограмм проводили с использованием электронной картотеки PCPDF – 95 и базы данных JCSD [15]. СТМ-изображения поверхности регистрировали на микроскопе LitScan2 [16].
![]() Как уже отмечалось, в подкисленных растворах вольфраматов возможно протекание нескольких конкурирующих процессов. На схеме 1 представлены некоторые взаимопревращения изополивольфраматов в подкисленном водном растворе. При рассмотрении этих взаимопревращений в литературе [4 – 8] принято использовать величины относительных кислотностей z = p / m, которые определяются стехиометрическим отношением количеств ионов гидроксония (р) и эквивалентов WO42- (m), необходимых для образования того или иного полианиона. В исследуемых растворах z составляют порядка сотен, т.е. заведомо не могут отвечать существованию какого-либо растворимого ИПА в условиях равновесия (для этого требуются z не выше 1,5 – 2). В то же время существует возможность реализовать метастабильные истинные растворы, т.к. в некоторых условиях установление равновесий в исследуемой системе происходит очень медленно.
Как уже отмечалось, в подкисленных растворах вольфраматов возможно протекание нескольких конкурирующих процессов. На схеме 1 представлены некоторые взаимопревращения изополивольфраматов в подкисленном водном растворе. При рассмотрении этих взаимопревращений в литературе [4 – 8] принято использовать величины относительных кислотностей z = p / m, которые определяются стехиометрическим отношением количеств ионов гидроксония (р) и эквивалентов WO42- (m), необходимых для образования того или иного полианиона. В исследуемых растворах z составляют порядка сотен, т.е. заведомо не могут отвечать существованию какого-либо растворимого ИПА в условиях равновесия (для этого требуются z не выше 1,5 – 2). В то же время существует возможность реализовать метастабильные истинные растворы, т.к. в некоторых условиях установление равновесий в исследуемой системе происходит очень медленно.
![]() Три формы представленных на схеме 1 изополивольфраматов являются спектрофотометрически активными в УФ-области: W7 (плато в области 260 нм [17]), W10 (максимум в области 320 нм [18]) и a-W12 (максимум в области 260 нм [19]), поэтому для качественного контроля состояния растворов использовали спектрофотометрический метод.
Три формы представленных на схеме 1 изополивольфраматов являются спектрофотометрически активными в УФ-области: W7 (плато в области 260 нм [17]), W10 (максимум в области 320 нм [18]) и a-W12 (максимум в области 260 нм [19]), поэтому для качественного контроля состояния растворов использовали спектрофотометрический метод.
![]() На рис. 1 представлены серии спектров поглощения 0,02 мМэ растворов мономерного вольфрамата, которые готовили непосредственно перед каждым измерением из раствора с концентрацией 1 мМэ через 10 – 90 минут после его приготовления. Для аналогичных растворов на основе паравольфрамата наблюдается качественно близкая форма спектров: ступень в области ~ 260 нм и плечо или максимум при ~ 320 нм, что можно рассматривать как свидетельство присутствия в растворе полимерных форм W7 и W10. Таким образом, независимо от степени полимеризации исходного твердого соединения, вводимого в раствор, через некоторое время составы растворов сближаются.
На рис. 1 представлены серии спектров поглощения 0,02 мМэ растворов мономерного вольфрамата, которые готовили непосредственно перед каждым измерением из раствора с концентрацией 1 мМэ через 10 – 90 минут после его приготовления. Для аналогичных растворов на основе паравольфрамата наблюдается качественно близкая форма спектров: ступень в области ~ 260 нм и плечо или максимум при ~ 320 нм, что можно рассматривать как свидетельство присутствия в растворе полимерных форм W7 и W10. Таким образом, независимо от степени полимеризации исходного твердого соединения, вводимого в раствор, через некоторое время составы растворов сближаются.
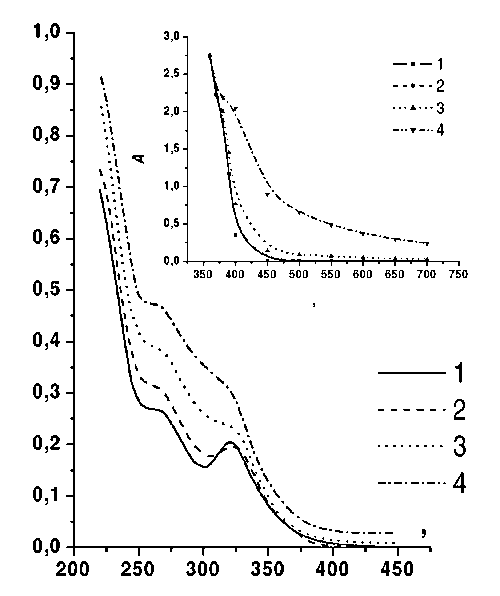 |
| Рис. 1. Спектры поглощения в УФ области раствора 1мМэ Na2WO4 + 0,5 М H2SO4, разбавленного (перед каждым измерением) до концентрации 0,02 мМэ. На врезке - динамика изменения оптических спектров поглощения того же раствора (без разбавления) в видимой области. Время после приготовления, мин: 1 – 10, 2 – 50, 3 – 90, 4 – 150. |
![]() В принципе, величины поглощения на характеристических участках спектров могли бы быть использованы для оценки содержания различных форм ИПА в растворе. Однако выполнить такой анализ на количественном уровне не представляется возможным, поскольку в растворах вольфраматов всегда реализуются равновесия с участием нескольких форм, и спектры, соответствующие присутствию только одной формы (W10 или W7), в литературе отсутствуют. В любом случае невозможно определить содержание спектрофотометрически неактивных форм. Ограничимся перечислением некоторых тенденций.
В принципе, величины поглощения на характеристических участках спектров могли бы быть использованы для оценки содержания различных форм ИПА в растворе. Однако выполнить такой анализ на количественном уровне не представляется возможным, поскольку в растворах вольфраматов всегда реализуются равновесия с участием нескольких форм, и спектры, соответствующие присутствию только одной формы (W10 или W7), в литературе отсутствуют. В любом случае невозможно определить содержание спектрофотометрически неактивных форм. Ограничимся перечислением некоторых тенденций.
![]() (1) При старении раствора, приготовленного из мономерного вольфрамата, наблюдается увеличение оптической плотности при 260 нм и, при небольших временах, уменьшение высоты пика при 320 нм (рис. 1). Такое поведение растворов вольфрамата можно рассматривать как следствие установления равновесия между гептавольфраматом и декавольфраматом, хотя не исключено, что эти равновесия носят более сложный характер и протекают в несколько стадий.
(1) При старении раствора, приготовленного из мономерного вольфрамата, наблюдается увеличение оптической плотности при 260 нм и, при небольших временах, уменьшение высоты пика при 320 нм (рис. 1). Такое поведение растворов вольфрамата можно рассматривать как следствие установления равновесия между гептавольфраматом и декавольфраматом, хотя не исключено, что эти равновесия носят более сложный характер и протекают в несколько стадий.
![]() (2) Из соотношения высот плато при 260 и максимума или плато при 320 нм можно заключить, что соотношение W7/W10 выше в растворе, приготовленном из W12.
(2) Из соотношения высот плато при 260 и максимума или плато при 320 нм можно заключить, что соотношение W7/W10 выше в растворе, приготовленном из W12.
![]() (3) В случае раствора паравольфрамата, согласно приведенному в [4] значению константы равновесия, в присутствии 0,5 М H2SO4 равновесие должно быть смещено в сторону образования W12, и додекамерной формы в растворе должно быть в ~ 16 раз больше, чем гептамерной. Однако в исследуемом нами растворе это равновесие явно нарушается ввиду присутствия формы W10.
(3) В случае раствора паравольфрамата, согласно приведенному в [4] значению константы равновесия, в присутствии 0,5 М H2SO4 равновесие должно быть смещено в сторону образования W12, и додекамерной формы в растворе должно быть в ~ 16 раз больше, чем гептамерной. Однако в исследуемом нами растворе это равновесие явно нарушается ввиду присутствия формы W10.
![]() (4) В обоих растворах вряд ли имеются заметные количества мономерной формы, в противном случае происходило бы быстрое образование коллоида H2WO4.
(4) В обоих растворах вряд ли имеются заметные количества мономерной формы, в противном случае происходило бы быстрое образование коллоида H2WO4.
![]() (5) Поскольку на спектре не наблюдается максимума в области 260 нм, можно считать, что образования метавольфрамата (a-W12) в заметных количествах не происходит. Исключить присутствие других полимерных форм вольфрамата в растворах из моно- и паравольфрамата нельзя.
(5) Поскольку на спектре не наблюдается максимума в области 260 нм, можно считать, что образования метавольфрамата (a-W12) в заметных количествах не происходит. Исключить присутствие других полимерных форм вольфрамата в растворах из моно- и паравольфрамата нельзя.
![]() На врезке рис. 1 представлена серия кривых в видимой области спектра, измеренная без разбавления исходного раствора. Видно, что при выбранной методике растворения образование коллоида (признаком которого является слабо зависящее от длины волны поглощение в широкой спектральной области) обнаруживаются не ранее чем через 70 – 90 минут после приготовления раствора, что позволяет завершить потенциодинамические измерения до начала образования коллоида. Отметим, что при внесении навесок вольфраматов в раствор 0,5 М Н2SO4 при комнатной температуре полного растворения не достигается, а образование коллоидной вольфрамовой кислоты визуально фиксируется уже в первые минуты. Все представленные ниже электрохимические данные относятся только к растворам, в которых не обнаруживалось поглощения в видимой области.
На врезке рис. 1 представлена серия кривых в видимой области спектра, измеренная без разбавления исходного раствора. Видно, что при выбранной методике растворения образование коллоида (признаком которого является слабо зависящее от длины волны поглощение в широкой спектральной области) обнаруживаются не ранее чем через 70 – 90 минут после приготовления раствора, что позволяет завершить потенциодинамические измерения до начала образования коллоида. Отметим, что при внесении навесок вольфраматов в раствор 0,5 М Н2SO4 при комнатной температуре полного растворения не достигается, а образование коллоидной вольфрамовой кислоты визуально фиксируется уже в первые минуты. Все представленные ниже электрохимические данные относятся только к растворам, в которых не обнаруживалось поглощения в видимой области.
![]() При сравнении потенциодинамических кривых, полученных в растворах изополивольфраматов различной концентрации, с кривыми в растворе фона можно отметить несколько очевидных различий (рис. 2).
При сравнении потенциодинамических кривых, полученных в растворах изополивольфраматов различной концентрации, с кривыми в растворе фона можно отметить несколько очевидных различий (рис. 2).
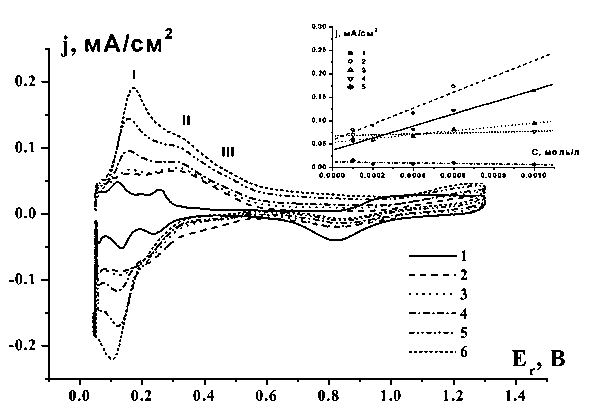 |
| Рис. 2. Потенциодинамические кривые гладкого платинового электрода в растворах с различной концентрацией (Мэ) Na2WO4 в 0,5 М H2SO4 при скорости развертки v = 20 мВ/с. 1 - раствор фона 0,5 М H2SO4, 2 – 1*10-4, 3 – 2*10-4, 4 – 4*10-4, 5 – 6*10-4, 6 – 1*10-3. На врезке зависимость токов в характерных интервалах потенциала от концентрации: 1, 2, 5 – для растворов Na2WO4, интервалы I, II и III соответственно; 3, 4 – интервалы I и II в растворах (NH4)10[H2W12O42]. |
![]() Во-первых, при добавлении вольфрамата в раствор резко изменяется форма кривых и увеличивается заряд в водородной области, причем тем сильнее, чем выше концентрация добавки. В миллимолярных растворах обсуждаемые заряды в 4 – 6 раз превышают затраты заряда на десорбцию монослоя водорода. Можно заключить, что одновременно с десорбцией водорода на анодном ходе кривой происходит окисление восстановленной формы вольфрамата, накопленной на катодном ходе и во время задержки при потенциале 0,05 В, причем продукт восстановления может находиться как в приэлектродном слое, так и на поверхности электрода.
Во-первых, при добавлении вольфрамата в раствор резко изменяется форма кривых и увеличивается заряд в водородной области, причем тем сильнее, чем выше концентрация добавки. В миллимолярных растворах обсуждаемые заряды в 4 – 6 раз превышают затраты заряда на десорбцию монослоя водорода. Можно заключить, что одновременно с десорбцией водорода на анодном ходе кривой происходит окисление восстановленной формы вольфрамата, накопленной на катодном ходе и во время задержки при потенциале 0,05 В, причем продукт восстановления может находиться как в приэлектродном слое, так и на поверхности электрода.
![]() Второе различие связано с изменениями хода кривых в двойнослойной области. Наблюдается существенное увеличение тока на анодном ходе при увеличении концентрации вольфрамата (рис. 2). На катодном ходе потенциодинамической кривой, напротив, токи становятся меньше, а при высоких концентрациях добавки даже изменяют знак в некотором интервале потенциалов. Это можно объяснить окислением восстановленного вольфрамата, которое вносит вклад в ток, как на анодном, так и на катодном ходе кривых.
Второе различие связано с изменениями хода кривых в двойнослойной области. Наблюдается существенное увеличение тока на анодном ходе при увеличении концентрации вольфрамата (рис. 2). На катодном ходе потенциодинамической кривой, напротив, токи становятся меньше, а при высоких концентрациях добавки даже изменяют знак в некотором интервале потенциалов. Это можно объяснить окислением восстановленного вольфрамата, которое вносит вклад в ток, как на анодном, так и на катодном ходе кривых.
![]() В третьих, в растворах вольфраматов заряд в кислородной области значительно меньше, чем в растворе фона, и мало зависит от концентрации вольфрамата.
В третьих, в растворах вольфраматов заряд в кислородной области значительно меньше, чем в растворе фона, и мало зависит от концентрации вольфрамата.
![]() Все перечисленные особенности имеют место и в растворах, приготовленных из паравольфрамата, хотя существуют некоторые количественные различия.
Все перечисленные особенности имеют место и в растворах, приготовленных из паравольфрамата, хотя существуют некоторые количественные различия.
![]() В водородной области обсуждаемых кривых можно выделить три пары размытых пиков или плато в интервалах потенциалов, обозначенных на рис. 2 I , II и III. Дальнейший анализ зависимостей токов от концентрации добавки и скорости развертки потенциала направлен на определение природы процессов, протекающих в соответствующих интервалах потенциалов.
В водородной области обсуждаемых кривых можно выделить три пары размытых пиков или плато в интервалах потенциалов, обозначенных на рис. 2 I , II и III. Дальнейший анализ зависимостей токов от концентрации добавки и скорости развертки потенциала направлен на определение природы процессов, протекающих в соответствующих интервалах потенциалов.
![]() На врезке к рис. 2 приведены зависимости анодных токов при фиксированном потенциале в интервалах I и II от концентрации растворов, приготовленных из Na2WO4 (кривые 1, 2) и из (NH4)10[H2W12O42] (кривые 3, 4). Поправка на вклад токов в отсутствие вольфраматов внесена вычитанием тока в растворе фона. Токи в обоих случаях практически линейно увеличиваются с концентрацией, но ни одна из приведенных зависимостей не стремится в ноль при нулевой концентрации, что является свидетельством наложения откликов нескольких процессов. Поскольку внесенная поправка скорее всего переоценена, можно утверждать, что значительный вклад в ток вносит некий процесс с участием вольфрамата, скорость которого не зависит от концентрации добавки. Для катодных токов в интервале III (кривая 5 рис. 2) с поправкой на постоянный заряд в двойнослойной области зависимость от концентрации добавки в исследованном интервале отсутствует.
На врезке к рис. 2 приведены зависимости анодных токов при фиксированном потенциале в интервалах I и II от концентрации растворов, приготовленных из Na2WO4 (кривые 1, 2) и из (NH4)10[H2W12O42] (кривые 3, 4). Поправка на вклад токов в отсутствие вольфраматов внесена вычитанием тока в растворе фона. Токи в обоих случаях практически линейно увеличиваются с концентрацией, но ни одна из приведенных зависимостей не стремится в ноль при нулевой концентрации, что является свидетельством наложения откликов нескольких процессов. Поскольку внесенная поправка скорее всего переоценена, можно утверждать, что значительный вклад в ток вносит некий процесс с участием вольфрамата, скорость которого не зависит от концентрации добавки. Для катодных токов в интервале III (кривая 5 рис. 2) с поправкой на постоянный заряд в двойнослойной области зависимость от концентрации добавки в исследованном интервале отсутствует.
![]() На рис. 3 приведен пример зависимости хода потенциодинамических кривых от скорости развертки потенциала. Токи в интервалах потенциалов I и II в водородной области по-разному зависят от скорости развертки v. В частности, для интервала II увеличение тока с ростом v происходит более резко, чем это предсказывается для процессов с участием растворенных частиц при диффузионном контроле (кривая 2 врезки к рис. 3). Анализ близких к линейным (по крайней мере, для анодных токов) зависимостей lg j – lg v позволяет предположить одновременное протекание процессов разных типов, для которых определенные по наклону степенные коэффициенты составляют 0,5 и 1, причем для токов в интервале I обсуждаемые коэффициенты ненамного выше 0,5, а для токов плато в интервале II они всегда существенно выше и в ряде случаев близки к 1. Последнее указывает на преимущественный вклад процессов, парциальные токи которых линейно зависят от скорости развертки.
На рис. 3 приведен пример зависимости хода потенциодинамических кривых от скорости развертки потенциала. Токи в интервалах потенциалов I и II в водородной области по-разному зависят от скорости развертки v. В частности, для интервала II увеличение тока с ростом v происходит более резко, чем это предсказывается для процессов с участием растворенных частиц при диффузионном контроле (кривая 2 врезки к рис. 3). Анализ близких к линейным (по крайней мере, для анодных токов) зависимостей lg j – lg v позволяет предположить одновременное протекание процессов разных типов, для которых определенные по наклону степенные коэффициенты составляют 0,5 и 1, причем для токов в интервале I обсуждаемые коэффициенты ненамного выше 0,5, а для токов плато в интервале II они всегда существенно выше и в ряде случаев близки к 1. Последнее указывает на преимущественный вклад процессов, парциальные токи которых линейно зависят от скорости развертки.
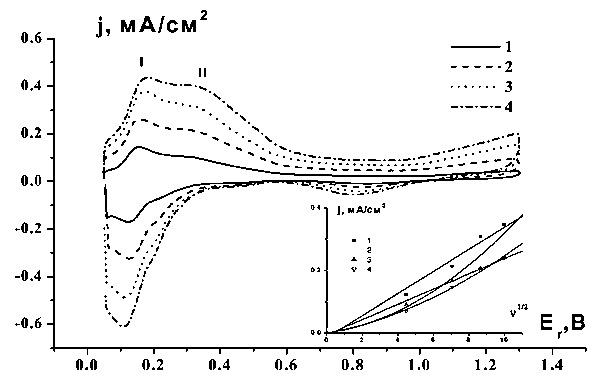 |
| Рис. 3. Потенциодинамические кривые гладкого платинового электрода в растворе 6*10-4 Мэ Na2WO4 в 0,5 М H2SO4 при разных скоростях развертки v, мВ/с: 1 – 20, 2 – 50, 3 – 75, 4 – 100. На врезке зависимость токов на характерных интервалах кривых от скорости развертки: 1, 2 – для раствора Na2WO4, интервалы I и II соответственно; 3, 4 – интервалы I и II в растворе (NH4)10[H2W12O42]. |
![]() На основании данных рис. 2 и 3 можно, таким образом, предположить протекание в системе параллельных процессов с участием растворенных вольфраматных частиц (контролируется диффузией,
На основании данных рис. 2 и 3 можно, таким образом, предположить протекание в системе параллельных процессов с участием растворенных вольфраматных частиц (контролируется диффузией, ![]() ) и с участием редокс-активного адсорбата или перезаряжаемой пленки (
) и с участием редокс-активного адсорбата или перезаряжаемой пленки (![]() ). Обнаружение первого из указанных процессов достаточно неожиданно. Действительно, как обсуждалось выше, потенциалы первого одноэлектронного редокс-перехода для кислых растворов вольфраматов как при рН
). Обнаружение первого из указанных процессов достаточно неожиданно. Действительно, как обсуждалось выше, потенциалы первого одноэлектронного редокс-перехода для кислых растворов вольфраматов как при рН ![]() 0 [11 – 13], так и при рН 4 – 6 заведомо отрицательнее обратимого водородного. По-видимому, потенциалы [11 – 13] следует относить не к ИПА, а к образующимся в присутствии избытка НСl хлоридным комплексам типа WO2Cl2.
0 [11 – 13], так и при рН 4 – 6 заведомо отрицательнее обратимого водородного. По-видимому, потенциалы [11 – 13] следует относить не к ИПА, а к образующимся в присутствии избытка НСl хлоридным комплексам типа WO2Cl2.
![]() В то же время трудно ожидать принципиальных различий состава лигандного окружения в исследованных сернокислых растворах и в ацетатных буферных растворах [14]. Следует предположить, что расхождение с данными [14] обусловлены различиями кинетики восстановления: на стеклоуглероде при рН 4 – 6 процесс протекает значительно медленнее, чем на Pt при рН 0,5. Это предположение согласуется с сильными различиями формы вольтамперограмм (приведенные в [14] кривые заведомо необратимы, а при рН 6 пик на обратном ходе в [14] вообще не удалось зарегистрировать). Отмеченные различия в кинетике восстановления могут быть связаны как с рН-зависимостью скорости какой-либо стадии, так и с различиями в адсорбции реагентов на платине и стеклоуглероде.
В то же время трудно ожидать принципиальных различий состава лигандного окружения в исследованных сернокислых растворах и в ацетатных буферных растворах [14]. Следует предположить, что расхождение с данными [14] обусловлены различиями кинетики восстановления: на стеклоуглероде при рН 4 – 6 процесс протекает значительно медленнее, чем на Pt при рН 0,5. Это предположение согласуется с сильными различиями формы вольтамперограмм (приведенные в [14] кривые заведомо необратимы, а при рН 6 пик на обратном ходе в [14] вообще не удалось зарегистрировать). Отмеченные различия в кинетике восстановления могут быть связаны как с рН-зависимостью скорости какой-либо стадии, так и с различиями в адсорбции реагентов на платине и стеклоуглероде.
![]() Все приведенные выше данные относятся к начальным циклам, зарегистрированным в каждом из исследуемых растворов (это, как правило, 4 – 8-ой циклы). При более длительном циклировании происходит увеличение тока, свидетельствующее о накоплении редокс-активного вещества на поверхности (рис. 4). С целью независимой характеристики этого продукта были проведены эксперименты с отмывкой электрода, позволяющие исключить вклад процессов с участием растворенного вольфрамата.
Все приведенные выше данные относятся к начальным циклам, зарегистрированным в каждом из исследуемых растворов (это, как правило, 4 – 8-ой циклы). При более длительном циклировании происходит увеличение тока, свидетельствующее о накоплении редокс-активного вещества на поверхности (рис. 4). С целью независимой характеристики этого продукта были проведены эксперименты с отмывкой электрода, позволяющие исключить вклад процессов с участием растворенного вольфрамата.
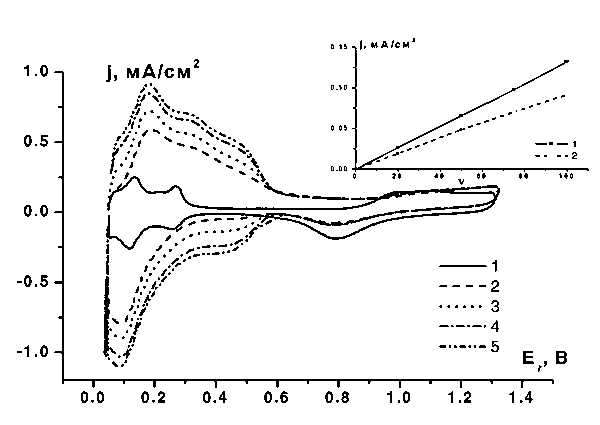 |
| Рис. 4. Серия потенциодинамических кривых, полученных при циклировании потенциала от 0,05 до 1,3 В в 1 мМэ растворе Na2WO4, v = 100 мВ/с. 1 – в растворе фона 0,5 М H2SO4, 2 – 7-й, 3 – 20-й, 4 – 40-й, 5 – 53-й цикл. На врезке зависимость токов от скорости развертки для участка III: 1 – Na2WO4, 2 – (NH4)10[H2W12O42]. |
![]() Постепенное увеличение заряда в интервале потенциалов 0,05 – 0,6 В как на анодном, так и на катодном ходе кривых (рис. 4) удается проследить в ходе циклирования в течение приблизительно 40 – 60 мин.. Приращение заряда от цикла к циклу при этом приблизительно постоянно, т.е. нет оснований считать, что какие-либо факторы ограничивают рост пленок при увеличении их толщины. Более длительное циклирование невозможно из-за ограниченного времени жизни неравновесных истинных растворов (см. выше).
Постепенное увеличение заряда в интервале потенциалов 0,05 – 0,6 В как на анодном, так и на катодном ходе кривых (рис. 4) удается проследить в ходе циклирования в течение приблизительно 40 – 60 мин.. Приращение заряда от цикла к циклу при этом приблизительно постоянно, т.е. нет оснований считать, что какие-либо факторы ограничивают рост пленок при увеличении их толщины. Более длительное циклирование невозможно из-за ограниченного времени жизни неравновесных истинных растворов (см. выше).
![]() Сопоставление потенциодинамических кривых до и после отмывки (кривые 2 и 3 рис. 5) показывает, что смещение токов в двойнослойной области в растворе вольфрамата связано исключительно с процессами, протекающими с участием растворенных вольфраматных частиц. В растворе фона после модификации электрода и отмывки двойнослойная область, по крайней мере при Еr > 0,6 В, совпадает с аналогичной областью на кривой чистого электрода (кривые 3 и 1 рис. 5 соответственно). В результате отмывки увеличивается также заряд в кислородной области, оставаясь, однако, несколько меньше, чем заряд на кривой для чистого электрода в растворе фона.
Сопоставление потенциодинамических кривых до и после отмывки (кривые 2 и 3 рис. 5) показывает, что смещение токов в двойнослойной области в растворе вольфрамата связано исключительно с процессами, протекающими с участием растворенных вольфраматных частиц. В растворе фона после модификации электрода и отмывки двойнослойная область, по крайней мере при Еr > 0,6 В, совпадает с аналогичной областью на кривой чистого электрода (кривые 3 и 1 рис. 5 соответственно). В результате отмывки увеличивается также заряд в кислородной области, оставаясь, однако, несколько меньше, чем заряд на кривой для чистого электрода в растворе фона.
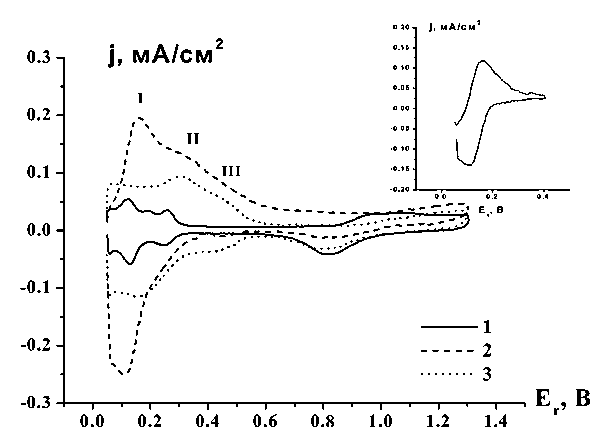 |
| Рис. 5. Серия потенциодинамических кривых гладкого платинового электрода в опытах с отмывкой. Скорость развертки v = 20 мВ/с. 1 – кривая в растворе фона 0,5 М H2SO4 до модификации электрода, 2 – кривая в 0,5 М H2SO4 растворе 1мМэ Na2WO4, 3 - кривая в растворе фона 0,5 М H2SO4 после модификации электрода (~ 10 циклов). На врезке представлен результат вычитания токов, регистрируемых после отмывки электрода (3) из токов в растворе вольфрамата (2). |
![]() В то же время в водородной области заряд на модифицированном электроде после отмывки по-прежнему значительно выше, чем на чистой поверхности. По сравнению с кривой в растворе вольфрамата токи в интервалах II и III в водородной области изменяются в результате отмывки значительно меньше, чем высота пика в интервале I. Таким образом, основная часть заряда, отвечающего интервалам II и III, относится к редокс-превращениям вещества, не удаляющегося при отмывке.
В то же время в водородной области заряд на модифицированном электроде после отмывки по-прежнему значительно выше, чем на чистой поверхности. По сравнению с кривой в растворе вольфрамата токи в интервалах II и III в водородной области изменяются в результате отмывки значительно меньше, чем высота пика в интервале I. Таким образом, основная часть заряда, отвечающего интервалам II и III, относится к редокс-превращениям вещества, не удаляющегося при отмывке.
![]() В области потенциалов I после отмывки наблюдается плато, высота которого не слишком сильно отличается от высоты пика десорбции слабосвязанного водорода на чистом электроде. Такое существенное уменьшение высот анодного (Еr = 0,167 В) и катодного (Еr = 0,110 В) пиков после отмывки электрода позволяет отнести эти пики к редокс-превращениям с участием растворенных частиц.
В области потенциалов I после отмывки наблюдается плато, высота которого не слишком сильно отличается от высоты пика десорбции слабосвязанного водорода на чистом электроде. Такое существенное уменьшение высот анодного (Еr = 0,167 В) и катодного (Еr = 0,110 В) пиков после отмывки электрода позволяет отнести эти пики к редокс-превращениям с участием растворенных частиц.
![]() В области потенциалов 0,4 – 0,6 В на анодном ходе наблюдается линейная зависимость токов от скорости развертки (врезка к рис. 4), причем при всех потенциалах экстраполяция к v = 0 дает нулевые значения тока. Величины катодного и анодного зарядов в водородной области на кривой после отмывки практически равны. Таким образом, в растворах вольфраматов образуется (и не удаляется при отмывке) перезаряжаемая пленка, характеризующаяся достаточно высокой скоростью транспорта заряда.
В области потенциалов 0,4 – 0,6 В на анодном ходе наблюдается линейная зависимость токов от скорости развертки (врезка к рис. 4), причем при всех потенциалах экстраполяция к v = 0 дает нулевые значения тока. Величины катодного и анодного зарядов в водородной области на кривой после отмывки практически равны. Таким образом, в растворах вольфраматов образуется (и не удаляется при отмывке) перезаряжаемая пленка, характеризующаяся достаточно высокой скоростью транспорта заряда.
![]() Обратимое заряжение пленки связано, по-видимому, с изменением кислородной стехиометрии. В качестве материала – аналога можно упомянуть, например, пленку оксида, химически осажденного из раствора WCl6 в изопропаноле (толщина полученной пленки составляла около 200 нм, т.е. ~ 250 монослоев) [20]. По данным in situ ИК- спектроскопии, полученным в растворе 0,5 M H2SO4 в интервале потенциалов -0,2
Обратимое заряжение пленки связано, по-видимому, с изменением кислородной стехиометрии. В качестве материала – аналога можно упомянуть, например, пленку оксида, химически осажденного из раствора WCl6 в изопропаноле (толщина полученной пленки составляла около 200 нм, т.е. ~ 250 монослоев) [20]. По данным in situ ИК- спектроскопии, полученным в растворе 0,5 M H2SO4 в интервале потенциалов -0,2 ![]() 1,0 В по н.к.э. в частотном диапазоне 500 – 4000 см-1, восстановление такой пленки (переход WVI/WV) сопровождается протонированием и, соответственно, увеличением числа связей О–Н. Схематически в [20] процесс заряжения представлен следующим образом:
1,0 В по н.к.э. в частотном диапазоне 500 – 4000 см-1, восстановление такой пленки (переход WVI/WV) сопровождается протонированием и, соответственно, увеличением числа связей О–Н. Схематически в [20] процесс заряжения представлен следующим образом:
| xH+(р-р) | (1) |
| O2WVI=O +xe- | (2) |
| O2WV–Ox- + xH+(абс) | (3) |
![]() Продукт реакции (3) представляет собой водородвольфрамовую бронзу. При ее окислении обратный процесс приводит к уменьшению числа связей О–Н в пленке. Полученные в [20] ИК-спектры подтверждают включение в структуру пленки воды при восстановлении и образование гидратированного нестехиометрического оксида:
Продукт реакции (3) представляет собой водородвольфрамовую бронзу. При ее окислении обратный процесс приводит к уменьшению числа связей О–Н в пленке. Полученные в [20] ИК-спектры подтверждают включение в структуру пленки воды при восстановлении и образование гидратированного нестехиометрического оксида:
| O2WVI–О + xH3О+(р-р) + xe- | (4) |
![]() Благодаря способности протона диффундировать в объем пленки вольфрамовые бронзы оказываются способными к довольно быстрому высокообратимому перезаряжению. Число различных материалов этого типа очень велико, они активно исследуются в последние годы в связи с электрохромными приложениями [21].
Благодаря способности протона диффундировать в объем пленки вольфрамовые бронзы оказываются способными к довольно быстрому высокообратимому перезаряжению. Число различных материалов этого типа очень велико, они активно исследуются в последние годы в связи с электрохромными приложениями [21].
![]() Для некоторых таких материалов типично наличие двух пиков в водородной области. Согласно [22] эти отклики связаны с образованием двух форм водородвольфрамовых бронз. Формально соответствующие им процессы можно представить уравнениями:
Для некоторых таких материалов типично наличие двух пиков в водородной области. Согласно [22] эти отклики связаны с образованием двух форм водородвольфрамовых бронз. Формально соответствующие им процессы можно представить уравнениями:
| WO3 + 0,18H + + 0,18e- | (5) |
| H0,18WO3 + 0,17H + + 0,17e- | (6) |
![]() В [10, 22] отмечают, что для пленок HxWO3, соосажденных с Pt на стеклоуглероде, характерно наличие двух пиков, потенциалы которых зависят от температуры. При 24 oС в 2 M H2SO4 они равны 0,13 и -0,21 В (н.к.э.), при более высокой температуре 35 oС пики смещаются в область более отрицательных значений (-0,040 и -0,240 В (н.к.э.) соответственно).
В [10, 22] отмечают, что для пленок HxWO3, соосажденных с Pt на стеклоуглероде, характерно наличие двух пиков, потенциалы которых зависят от температуры. При 24 oС в 2 M H2SO4 они равны 0,13 и -0,21 В (н.к.э.), при более высокой температуре 35 oС пики смещаются в область более отрицательных значений (-0,040 и -0,240 В (н.к.э.) соответственно).
![]() По значительной разности зарядов водородных областей кривых 1 и 3 рис. 5 можно сделать заключение о полислойном характере электрохимически активной вольфраматной пленки на поверхности платины. Затраты заряда на процессы в пленке увеличиваются при увеличении времени циклирования в растворах с добавками вольфраматов, тогда как вклад процессов с участием растворенных частиц остается примерно постоянным.
По значительной разности зарядов водородных областей кривых 1 и 3 рис. 5 можно сделать заключение о полислойном характере электрохимически активной вольфраматной пленки на поверхности платины. Затраты заряда на процессы в пленке увеличиваются при увеличении времени циклирования в растворах с добавками вольфраматов, тогда как вклад процессов с участием растворенных частиц остается примерно постоянным.
![]() По зарядам водородной области на кривых в растворах изополивольфраматов можно провести предварительную оценку числа монослоев, которые удалось сформировать на поверхности электрода при максимально возможных в наших опытах временах осаждения. Ниже при оценке принято предположение о том, что образовавшаяся пленка представляет собой нестехиометрический гидратированный оксид вольфрама, поскольку область потенциалов, в которой проявляется редокс-активность пленки, очень близка к описанным в литературе именно для материалов такой природы.
По зарядам водородной области на кривых в растворах изополивольфраматов можно провести предварительную оценку числа монослоев, которые удалось сформировать на поверхности электрода при максимально возможных в наших опытах временах осаждения. Ниже при оценке принято предположение о том, что образовавшаяся пленка представляет собой нестехиометрический гидратированный оксид вольфрама, поскольку область потенциалов, в которой проявляется редокс-активность пленки, очень близка к описанным в литературе именно для материалов такой природы.
![]() На основании анализа, проведенного в [22, 23], можно предположить, что для полученного нами твердого продукта в исследуемой области потенциалов эффективно реализуется перенос 0,25 электрона, то есть изменение стехиометрии пленки от HxWO3 до Hx+0,25WO3. Примем, что на 1 см2 поверхности платины располагается 1,7*1014 частиц HxWO3 (оценка по параметру решетки 0,77 нм [24]). Тогда затраты заряда на редокс-превращения одного монослоя на площади 1 см2 должны составить 2,7*10-5 Кл. Для наиболее толстых из полученных пленок в рамках такой приближенной оценки число монослоев достигает ~ 400. Соответственно, толщина достаточно долго формировавшейся пленки составит 300 – 400 нм. Данные рис. 5 соответствуют пленке толщиной около 60 монослоев.
На основании анализа, проведенного в [22, 23], можно предположить, что для полученного нами твердого продукта в исследуемой области потенциалов эффективно реализуется перенос 0,25 электрона, то есть изменение стехиометрии пленки от HxWO3 до Hx+0,25WO3. Примем, что на 1 см2 поверхности платины располагается 1,7*1014 частиц HxWO3 (оценка по параметру решетки 0,77 нм [24]). Тогда затраты заряда на редокс-превращения одного монослоя на площади 1 см2 должны составить 2,7*10-5 Кл. Для наиболее толстых из полученных пленок в рамках такой приближенной оценки число монослоев достигает ~ 400. Соответственно, толщина достаточно долго формировавшейся пленки составит 300 – 400 нм. Данные рис. 5 соответствуют пленке толщиной около 60 монослоев.
![]() Данные опытов с отмывкой позволяют сделать также важное заключение о процессах с участием растворенных форм ИПА. На врезке рис. 5 показана кривая, полученная вычитанием кривой 3 из кривой 2 основного рис. 5. Она имеет ход, типичный для вольтамперограмм диффузионно контролируемого процесса с участием растворенных частиц. Разность потенциалов катодного и анодного пиков составляет около 60 мВ, что отвечает обратимому одноэлектронному процессу восстановления вольфрамата. Формальные потенциалы, определенные по среднему значению потенциалов катодного и анодного пиков, равны 0,142
Данные опытов с отмывкой позволяют сделать также важное заключение о процессах с участием растворенных форм ИПА. На врезке рис. 5 показана кривая, полученная вычитанием кривой 3 из кривой 2 основного рис. 5. Она имеет ход, типичный для вольтамперограмм диффузионно контролируемого процесса с участием растворенных частиц. Разность потенциалов катодного и анодного пиков составляет около 60 мВ, что отвечает обратимому одноэлектронному процессу восстановления вольфрамата. Формальные потенциалы, определенные по среднему значению потенциалов катодного и анодного пиков, равны 0,142![]() 0,05 и 0,128
0,05 и 0,128![]() 0,05 В для растворов, приготовленных из моно- и паравольфраматов соответственно. Этот результат согласуется со сделанным выше предположением о том, что формальные потенциалы, оцененные из необратимых вольтамперограмм [14], смещены от равновесного потенциала в сторону более отрицательных значений.
0,05 В для растворов, приготовленных из моно- и паравольфраматов соответственно. Этот результат согласуется со сделанным выше предположением о том, что формальные потенциалы, оцененные из необратимых вольтамперограмм [14], смещены от равновесного потенциала в сторону более отрицательных значений.
![]() Полученные разностные вольтамперограммы отвечают, вероятно, превращениям нескольких близких по редокс-свойствам форм ИПА, по крайней мере двух зафиксированных нами спектрофотометрически (W7 и W10). Незначительные различия формальных потенциалов, найденные для исследованных растворов, могут быть обусловлены разным соотношением в них указанных форм ИПА.
Полученные разностные вольтамперограммы отвечают, вероятно, превращениям нескольких близких по редокс-свойствам форм ИПА, по крайней мере двух зафиксированных нами спектрофотометрически (W7 и W10). Незначительные различия формальных потенциалов, найденные для исследованных растворов, могут быть обусловлены разным соотношением в них указанных форм ИПА.
![]() В дальнейшем заслуживает специального исследования вопрос о том, какую роль играет присутствие платиновой подложки в поведении перезаряжаемых пленок. В этой связи обращает на себя внимание тот факт, что область потенциалов, в которой наблюдается обратимое перезаряжение пленок на Pt, смещена в сторону более положительных значений по сравнению с аналогичной областью для водородвольфрамовых бронз на стеклоуглероде [22]. Согласно данным исследования вольфраматных оксидных пленок, соосажденных с платиной [25, 26], способность последней адсорбировать водород способствует образованию водородных оксидных бронз состава НxWO3.На потенциодинамических кривых, приведенных в [25], вместо водородной области, характерной для платины, наблюдается единственный пик в водородной области при потенциалах Er ~ 0,3 В, причем общий заряд водородной области больше, чем на платине, а в кислородной меньше. Качественно эта картина близка к наблюдаемой нами для пленок на гладкой платине. В [10] отмечают, что Pt, диспергированная в объеме WO3, смещает потенциал образования бронзы на 300 мВ (-0,240 В н.к.э.) по сравнению с аналогичным потенциалом для образцов оксида вольфрама, не содержащих примеси платины (-0,540 В н.к.э.). С учетом данных [10, 25] можно предположить, что в присутствии Рt перезаряжение вольфраматных пленок происходит по несколько иному механизму, чем (1) – (3). При потенциале Er > 0 начинается адсорбция водорода на платине (7). Адатомы водорода могут химически взаимодействовать с твердыми нестехиометрическими оксовольфраматами HxWO3 с образованием более восстановленной формы водородвольфрамовой бронзы Hx+dWO3 (8).
В дальнейшем заслуживает специального исследования вопрос о том, какую роль играет присутствие платиновой подложки в поведении перезаряжаемых пленок. В этой связи обращает на себя внимание тот факт, что область потенциалов, в которой наблюдается обратимое перезаряжение пленок на Pt, смещена в сторону более положительных значений по сравнению с аналогичной областью для водородвольфрамовых бронз на стеклоуглероде [22]. Согласно данным исследования вольфраматных оксидных пленок, соосажденных с платиной [25, 26], способность последней адсорбировать водород способствует образованию водородных оксидных бронз состава НxWO3.На потенциодинамических кривых, приведенных в [25], вместо водородной области, характерной для платины, наблюдается единственный пик в водородной области при потенциалах Er ~ 0,3 В, причем общий заряд водородной области больше, чем на платине, а в кислородной меньше. Качественно эта картина близка к наблюдаемой нами для пленок на гладкой платине. В [10] отмечают, что Pt, диспергированная в объеме WO3, смещает потенциал образования бронзы на 300 мВ (-0,240 В н.к.э.) по сравнению с аналогичным потенциалом для образцов оксида вольфрама, не содержащих примеси платины (-0,540 В н.к.э.). С учетом данных [10, 25] можно предположить, что в присутствии Рt перезаряжение вольфраматных пленок происходит по несколько иному механизму, чем (1) – (3). При потенциале Er > 0 начинается адсорбция водорода на платине (7). Адатомы водорода могут химически взаимодействовать с твердыми нестехиометрическими оксовольфраматами HxWO3 с образованием более восстановленной формы водородвольфрамовой бронзы Hx+dWO3 (8).
| Pt + H3O+ + e- | (7) |
| dPtH + HxWO3 | (8) |
![]() Разумеется, этот механизм справедлив для х = 0 (система Pt/WO3). Указанные процессы могут протекать и в отсутствие прямого контакта платины и восстановленного оксида, если адсорбированный на платине водород быстро диффундирует к HxWO3.
Разумеется, этот механизм справедлив для х = 0 (система Pt/WO3). Указанные процессы могут протекать и в отсутствие прямого контакта платины и восстановленного оксида, если адсорбированный на платине водород быстро диффундирует к HxWO3.
![]() При этом на свободной поверхности Pt высвобождаются места для дополнительной адсорбции водорода, а регистрируемый заряд отражает общее количество водорода, включая вступивший в реакцию с HxWO3. Такой механизм, аналогичный спилловер-эффекту, вполне реалистичен, по крайней мере для пленок малых толщин или высокодисперсных Рt-вольфрамоксидных композиций, т.е. в условиях значительной площади контакта металла с оксидом. Уточнение закономерностей транспорта заряда (в том числе протонов) в вольфраматных пленках требует специального исследования.
При этом на свободной поверхности Pt высвобождаются места для дополнительной адсорбции водорода, а регистрируемый заряд отражает общее количество водорода, включая вступивший в реакцию с HxWO3. Такой механизм, аналогичный спилловер-эффекту, вполне реалистичен, по крайней мере для пленок малых толщин или высокодисперсных Рt-вольфрамоксидных композиций, т.е. в условиях значительной площади контакта металла с оксидом. Уточнение закономерностей транспорта заряда (в том числе протонов) в вольфраматных пленках требует специального исследования.
![]() Рентгеновские дифрактограммы модифицированных паравольфраматом образцов поверхности показали наличие достаточно интенсивных рефлексов, не относящихся к откликам подложки, что однозначно подтверждает кристалличность вольфраматной пленки, образованной на поверхности. Идентифицировать фазы, образовавшиеся на поверхности, на данном этапе не удалось, однако по совпадению некоторых пиков с найденными в базе данных [15] фазами водородвольфрамовых бронз различного состава (H0,53WO3 (а), H0,5WO3 (б) H0,23WO3 (в), H0,66W1,64O5,23 (г)) можно предположить присутствие в пленке указанных компонентов. Так как некоторые из них отвечают более глубокому восстановлению WO3, чем предполагалось выше на основе сопоставления с данными для бронз на инертных подложках, выводы об изменении стехиометрии следует считать предварительными. Все перечисленные водородвольфрамовые бронзы – фазы, имеющие кубическую решетку с параметрами от 0,375 до 1,02 нм. Отметим, что приведенные данные относятся к извлеченным пленкам после хранения их на воздухе и не исключают однофазности пленок в растворе при фиксированном потенциале. Эксперименты в конфигурации СТМ подтверждают высокую проводимость образующихся пленок, природа проводимости требует дополнительных исследований. Сканирование оказывалось возможным уже при туннельном напряжении 0,2 В, что типично для материалов с проводимостью, близкой к металлической.
Рентгеновские дифрактограммы модифицированных паравольфраматом образцов поверхности показали наличие достаточно интенсивных рефлексов, не относящихся к откликам подложки, что однозначно подтверждает кристалличность вольфраматной пленки, образованной на поверхности. Идентифицировать фазы, образовавшиеся на поверхности, на данном этапе не удалось, однако по совпадению некоторых пиков с найденными в базе данных [15] фазами водородвольфрамовых бронз различного состава (H0,53WO3 (а), H0,5WO3 (б) H0,23WO3 (в), H0,66W1,64O5,23 (г)) можно предположить присутствие в пленке указанных компонентов. Так как некоторые из них отвечают более глубокому восстановлению WO3, чем предполагалось выше на основе сопоставления с данными для бронз на инертных подложках, выводы об изменении стехиометрии следует считать предварительными. Все перечисленные водородвольфрамовые бронзы – фазы, имеющие кубическую решетку с параметрами от 0,375 до 1,02 нм. Отметим, что приведенные данные относятся к извлеченным пленкам после хранения их на воздухе и не исключают однофазности пленок в растворе при фиксированном потенциале. Эксперименты в конфигурации СТМ подтверждают высокую проводимость образующихся пленок, природа проводимости требует дополнительных исследований. Сканирование оказывалось возможным уже при туннельном напряжении 0,2 В, что типично для материалов с проводимостью, близкой к металлической.
![]() Было проведено туннельно-микроскопическое сканирование достаточно толстых свежеосажденных (рис. 6) и состаренных (рис. 7) пленок. СТМ-изображения указывают на глобулярную структуру как свежеосажденных, так и состаренных осадков. На изображениях свежеосажденных пленок можно выделить ряды шириной ~ 100 нм (рис. 6а), состоящие из протяженных глобул толщиной 30 – 40 нм (рис. 6б). Глобулы имеют вытянутую форму и состоят из продольно расположенных протяженных фрагментов шириной не более 10 – 15 нм и длиной 20 – 30 нм (рис. 6в, г). Изображения состаренных пленок несколько отличаются от вышеописанных. На поверхности также можно различить ряды, ширина которых составляет 300 – 350 нм (рис. 7а). Глобулы имеют ширину 40 – 60 нм и длину 150 – 200 нм (рис. 7б). Структурные фрагменты, из которых состоят глобулы, расположены поперечно и имеют ширину 10 – 20 нм (рис. 7в). В обоих случаях удается визуализировать и более мелкие округлые структурные фрагменты с характерными размерами 5 – 10 нм, которые предположительно представляют собой единичные кубические кристаллы (рис. 7г) Эта предварительная морфологическая характеристика пленок позволяет говорить об их возможной дисперсности, способствующей, в частности, протонному транспорту. Можно также предположить, что процессы транспорта заряда в кристаллической решетке оказываются достаточно быстрыми благодаря малым размерам кристаллов.
Было проведено туннельно-микроскопическое сканирование достаточно толстых свежеосажденных (рис. 6) и состаренных (рис. 7) пленок. СТМ-изображения указывают на глобулярную структуру как свежеосажденных, так и состаренных осадков. На изображениях свежеосажденных пленок можно выделить ряды шириной ~ 100 нм (рис. 6а), состоящие из протяженных глобул толщиной 30 – 40 нм (рис. 6б). Глобулы имеют вытянутую форму и состоят из продольно расположенных протяженных фрагментов шириной не более 10 – 15 нм и длиной 20 – 30 нм (рис. 6в, г). Изображения состаренных пленок несколько отличаются от вышеописанных. На поверхности также можно различить ряды, ширина которых составляет 300 – 350 нм (рис. 7а). Глобулы имеют ширину 40 – 60 нм и длину 150 – 200 нм (рис. 7б). Структурные фрагменты, из которых состоят глобулы, расположены поперечно и имеют ширину 10 – 20 нм (рис. 7в). В обоих случаях удается визуализировать и более мелкие округлые структурные фрагменты с характерными размерами 5 – 10 нм, которые предположительно представляют собой единичные кубические кристаллы (рис. 7г) Эта предварительная морфологическая характеристика пленок позволяет говорить об их возможной дисперсности, способствующей, в частности, протонному транспорту. Можно также предположить, что процессы транспорта заряда в кристаллической решетке оказываются достаточно быстрыми благодаря малым размерам кристаллов.
 |
| Рис. 6. СТМ-изображения поверхности свежеприготовленной пленки. Размеры визуализированных участков: (а) – 361х346, (б) – 287,7х262,9, (в) – 178,6х166,1, (г) – 127,3х107,5 нм. |
 |
| Рис. 7. СТМ-изображения поверхности состаренной пленки. Размеры визуализированных участков: (а) – 639х635, (б) – 169х158,7, (в) – 120,1х133,6, (г) – 50,6х48,8 нм. |
![]() Признаков макроскопической несплошности пленок с толщинами около 400 монослоев (оцененными в предыдущем разделе) в СТМ-экспериментах не обнаружено, т.е. не зафиксировано участков с морфологией, характерной для поликристаллической платиновой фольги. Максимальные глубины, зарегистрированные в СТМ-экспериментах, составляют ~ 350 нм, что согласуется с оценками толщины по данным кулонометрии. В то же время, как следует из данных вольтамперометрии, адсорбция кислорода на платине даже при образовании столь толстых пленок не подавляется полностью. Это свидетельствует в пользу значительной пористости кристаллического осадка и его проницаемости для раствора. Следовательно, процессы перезаряжения нестехиометрического оксида могут инициироваться не только на наружной границе пленка – раствор, но и в порах.
Признаков макроскопической несплошности пленок с толщинами около 400 монослоев (оцененными в предыдущем разделе) в СТМ-экспериментах не обнаружено, т.е. не зафиксировано участков с морфологией, характерной для поликристаллической платиновой фольги. Максимальные глубины, зарегистрированные в СТМ-экспериментах, составляют ~ 350 нм, что согласуется с оценками толщины по данным кулонометрии. В то же время, как следует из данных вольтамперометрии, адсорбция кислорода на платине даже при образовании столь толстых пленок не подавляется полностью. Это свидетельствует в пользу значительной пористости кристаллического осадка и его проницаемости для раствора. Следовательно, процессы перезаряжения нестехиометрического оксида могут инициироваться не только на наружной границе пленка – раствор, но и в порах.
![]() Не детализируя механизма перезаряжения исследованных пленок, можно говорить о возможности реализации в них обратимых процессов общего вида:
Не детализируя механизма перезаряжения исследованных пленок, можно говорить о возможности реализации в них обратимых процессов общего вида:
| HxWO3 + dH+ + de- | (9) |
![]() Эти типичные для нестехиометрических гидратированных оксидов процессы, ранее наблюдавшиеся для многих систем и представляющие интерес в связи с созданием электрохимических конденсаторов [21], эффективных катализаторов окисления и восстановления [10, 22, 25, 26], высокоселективных сенсоров [24] и электрохромных устройств [20, 27 - 30], и т.д.
Эти типичные для нестехиометрических гидратированных оксидов процессы, ранее наблюдавшиеся для многих систем и представляющие интерес в связи с созданием электрохимических конденсаторов [21], эффективных катализаторов окисления и восстановления [10, 22, 25, 26], высокоселективных сенсоров [24] и электрохромных устройств [20, 27 - 30], и т.д.
![]() Выраженная кристалличность пленок и их морфология позволяют предположить, что образование твердых продуктов происходит в результате катодной электрокристаллизации. Благодаря более низкой растворимости восстановленных ИПА, высокая контролируемость процессов электрокристаллизации делает их перспективными для изготовления микро- и наноструктур, которые приобретают все большее значение в современной электронной технике. Особенно важное значение для этой задачи имеет осаждение фаз с переменной проводимостью, к которым относятся и вольфрамовые бронзы.
Выраженная кристалличность пленок и их морфология позволяют предположить, что образование твердых продуктов происходит в результате катодной электрокристаллизации. Благодаря более низкой растворимости восстановленных ИПА, высокая контролируемость процессов электрокристаллизации делает их перспективными для изготовления микро- и наноструктур, которые приобретают все большее значение в современной электронной технике. Особенно важное значение для этой задачи имеет осаждение фаз с переменной проводимостью, к которым относятся и вольфрамовые бронзы.
![]() Существенное влияние Рt на электрохимическое поведение нестехиометрических вольфраматов, известное по данным работ [10, 22, 26], указывает на перспективность материалов исследуемого типа для электрокатализа.
Существенное влияние Рt на электрохимическое поведение нестехиометрических вольфраматов, известное по данным работ [10, 22, 26], указывает на перспективность материалов исследуемого типа для электрокатализа.
![]() Мы благодарим Ф.М.Спиридонова за помощь в проведении исследования образцов методом рентгеновской дифракции и С.Ю. Васильева за помощь в проведении СТМ-экспериментов.
Мы благодарим Ф.М.Спиридонова за помощь в проведении исследования образцов методом рентгеновской дифракции и С.Ю. Васильева за помощь в проведении СТМ-экспериментов.
![]() Работа выполнена при финансовой поддержке РФФИ (грант 02-03-33285а).
Работа выполнена при финансовой поддержке РФФИ (грант 02-03-33285а).