![]() В материаловедении широко распространен общий подход к исследованию различных групп материалов, основанный на установлении взаимосвязей в ряду “синтез-состав-структура-свойство”. Однако все чаще обнаруживаются системы, в которых такой ряд в качестве промежуточного звена требует включения нового элемента, условно называемого “наноструктурой”. Именно такая ситуация имеет место, по-видимому, для многих интеркаляционных систем: транспорт интеркалята и равновесные сорбционные свойства оказываются существенно зависимыми не только от элементного и фазового состава матрицы и даже не только от ее дисперсности, но также от строения межзеренных границ и других фрагментов нанометровых размеров.
В материаловедении широко распространен общий подход к исследованию различных групп материалов, основанный на установлении взаимосвязей в ряду “синтез-состав-структура-свойство”. Однако все чаще обнаруживаются системы, в которых такой ряд в качестве промежуточного звена требует включения нового элемента, условно называемого “наноструктурой”. Именно такая ситуация имеет место, по-видимому, для многих интеркаляционных систем: транспорт интеркалята и равновесные сорбционные свойства оказываются существенно зависимыми не только от элементного и фазового состава матрицы и даже не только от ее дисперсности, но также от строения межзеренных границ и других фрагментов нанометровых размеров.
![]() Ярким примером являются компактированные под давлением наночастицы металлов [1,2]. Фундаментальные исследования нанокристаллического (н.к.) палладия (как порошков, так и компактов), выполненные с помощью рентгеноструктурного анализа, выявили необычные физические свойства этого материала – увеличенные по сравнению с крупнокристаллическим (к.к.) палладием статические атомные смещения, повышенную микродеформацию, малый размер областей когерентного рассеяния [3, 4]. Найденные различия свойств Pd(н.к.) и Pd(к.к.) объясняются возникновением неравновесных границ зерен с высоким уровнем внутренних напряжений и повышенной свободной энергией, обусловленных наличием зернограничных дислокаций [5 –7]. Для наноструктурированных палладиевых материалов такого типа (так называемых компактов) наблюдались различные аномалии в процессах сорбции водорода [8 - 10], исследованные для случаев образования a-и b фаз гидрида.
Ярким примером являются компактированные под давлением наночастицы металлов [1,2]. Фундаментальные исследования нанокристаллического (н.к.) палладия (как порошков, так и компактов), выполненные с помощью рентгеноструктурного анализа, выявили необычные физические свойства этого материала – увеличенные по сравнению с крупнокристаллическим (к.к.) палладием статические атомные смещения, повышенную микродеформацию, малый размер областей когерентного рассеяния [3, 4]. Найденные различия свойств Pd(н.к.) и Pd(к.к.) объясняются возникновением неравновесных границ зерен с высоким уровнем внутренних напряжений и повышенной свободной энергией, обусловленных наличием зернограничных дислокаций [5 –7]. Для наноструктурированных палладиевых материалов такого типа (так называемых компактов) наблюдались различные аномалии в процессах сорбции водорода [8 - 10], исследованные для случаев образования a-и b фаз гидрида.
![]() Электрохимические методы наноструктурирования [11] позволяют обеспечить неравновесные долгоживущие состояния материалов в значительно более мягких условиях (по сравнению с компактированием и лучевой обработкой). В отличие от других металлов, для палладия, наряду с величиной перенапряжения осаждения, в формировании специфических состояний электролитического осадка значительную роль играет сопутствующее (при некоторых режимах нанесения) гидридообразование. В частности, представляет значительный интерес возможность электроосаждения палладия при потенциалах образования b-фазы гидрида. Согласно [12 - 14], полученные в таких условиях образцы обладают сорбционной емкостью (атомное отношение Н/Pd>1) при эффективных давлениях водорода 0,1 – 1 атм, т.е. в условиях, в которых величина H/Pd для типичных палладиевых материалов не превышает 0,60-0,65. Менее выраженные аномалии демонстрируют и электролитические осадки Pd (ЭОPd), приготовленные при потенциалах образования a-фазы гидрида [12-16].
Электрохимические методы наноструктурирования [11] позволяют обеспечить неравновесные долгоживущие состояния материалов в значительно более мягких условиях (по сравнению с компактированием и лучевой обработкой). В отличие от других металлов, для палладия, наряду с величиной перенапряжения осаждения, в формировании специфических состояний электролитического осадка значительную роль играет сопутствующее (при некоторых режимах нанесения) гидридообразование. В частности, представляет значительный интерес возможность электроосаждения палладия при потенциалах образования b-фазы гидрида. Согласно [12 - 14], полученные в таких условиях образцы обладают сорбционной емкостью (атомное отношение Н/Pd>1) при эффективных давлениях водорода 0,1 – 1 атм, т.е. в условиях, в которых величина H/Pd для типичных палладиевых материалов не превышает 0,60-0,65. Менее выраженные аномалии демонстрируют и электролитические осадки Pd (ЭОPd), приготовленные при потенциалах образования a-фазы гидрида [12-16].
![]() Ранее аномальные палладиевые электролитические осадки были охарактеризованы рентгенографически [17, 18] и методом СТМ [12, 14]. Было показано, что средние размеры кристаллитов, визуализируемых в поверхностных слоях осадков, составляют 10 – 40 нм и зависят от потенциала осаждения Еd. Соответствующие распределения по размерам оказались достаточно широки, и их параметры также зависели от Еd. Оцененные для некоторых значений Еd периоды элементарной ячейки оказались ниже, чем для упорядоченной решетки массивного палладия. Были получены свидетельства в пользу наличия значительных макронапряжений в зернах ЭОPd. Настоящая работа посвящена более подробной рентгенографической и электроноспектроскопической характеристике аномальных электролитических осадков Рd и сопоставлению их особенностей с аналогичными, установленными ранее для других неравновесных материалов из палладия [3, 4]. В задачу исследования входит также выяснение вопроса о том, насколько состав и строение предшественников аномальных осадков – гидридов Pd , образующихся в области потенциалов a <=> b перехода, влияют на особенности кристаллического и электронного строения ЭОPd.
Ранее аномальные палладиевые электролитические осадки были охарактеризованы рентгенографически [17, 18] и методом СТМ [12, 14]. Было показано, что средние размеры кристаллитов, визуализируемых в поверхностных слоях осадков, составляют 10 – 40 нм и зависят от потенциала осаждения Еd. Соответствующие распределения по размерам оказались достаточно широки, и их параметры также зависели от Еd. Оцененные для некоторых значений Еd периоды элементарной ячейки оказались ниже, чем для упорядоченной решетки массивного палладия. Были получены свидетельства в пользу наличия значительных макронапряжений в зернах ЭОPd. Настоящая работа посвящена более подробной рентгенографической и электроноспектроскопической характеристике аномальных электролитических осадков Рd и сопоставлению их особенностей с аналогичными, установленными ранее для других неравновесных материалов из палладия [3, 4]. В задачу исследования входит также выяснение вопроса о том, насколько состав и строение предшественников аномальных осадков – гидридов Pd , образующихся в области потенциалов a <=> b перехода, влияют на особенности кристаллического и электронного строения ЭОPd.
![]() Палладий осаждали на платиновую фольгу из раствора 0,1 М PdCl2 + 1 M HCl при потенциалах Еd 0,026, 0,080 и 0,120 В относительно обратимого водородного электрода в растворе 0,5 М Н2SO4. Соответствующие образцы далее обозначены Pd26, Pd80 и Рd120.
Палладий осаждали на платиновую фольгу из раствора 0,1 М PdCl2 + 1 M HCl при потенциалах Еd 0,026, 0,080 и 0,120 В относительно обратимого водородного электрода в растворе 0,5 М Н2SO4. Соответствующие образцы далее обозначены Pd26, Pd80 и Рd120.
![]() Выбор потенциалов осаждения основан на данных [12] (в этой работе методика осаждения описана более подробно). Осадок Рd26 характеризуется рекордной сорбционной емкостью, он формируется в условиях образования b-фазы гидрида. Возможно, что процесс образования этого материала представляет собой непосредственно осаждение гидрида с его последующим разложением после извлечения образца. Осадки Pd80 и Рd120 образуются при одновременном образовании a-фазы гидрида, причем стационарная концентрация гидрида существенно выше для Рd80 (H/Pd порядка 0,01), чем для Рd120 (H/Pd порядка 0,001). Аномальные сорбционные свойства Pd80 и Рd120 проявляются в последующих опытах только в области образования a-фазы.
Выбор потенциалов осаждения основан на данных [12] (в этой работе методика осаждения описана более подробно). Осадок Рd26 характеризуется рекордной сорбционной емкостью, он формируется в условиях образования b-фазы гидрида. Возможно, что процесс образования этого материала представляет собой непосредственно осаждение гидрида с его последующим разложением после извлечения образца. Осадки Pd80 и Рd120 образуются при одновременном образовании a-фазы гидрида, причем стационарная концентрация гидрида существенно выше для Рd80 (H/Pd порядка 0,01), чем для Рd120 (H/Pd порядка 0,001). Аномальные сорбционные свойства Pd80 и Рd120 проявляются в последующих опытах только в области образования a-фазы.
![]() Для минимизации неравномерности осаждения вдоль поверхности использовалась ячейка с двумя параллельными платиновыми анодами большой площади, между которыми располагали электрод-подложку с площадью, меньшей в 5-6 раз. Для проверки воспроизводимости методики осаждения были сопоставлены рентгенограммы двух независимо приготовленных образцов Рd120. Различия рассчитанных для них величин параметра элементарной ячейки находились на верхней границе достигаемой погрешности (0,00006 нм). По-видимому, воспроизводимость наноструктуры не являлась полной, но различия были значительно меньше, чем найденные для образцов, полученных при разных потенциалах осаждения.
Для минимизации неравномерности осаждения вдоль поверхности использовалась ячейка с двумя параллельными платиновыми анодами большой площади, между которыми располагали электрод-подложку с площадью, меньшей в 5-6 раз. Для проверки воспроизводимости методики осаждения были сопоставлены рентгенограммы двух независимо приготовленных образцов Рd120. Различия рассчитанных для них величин параметра элементарной ячейки находились на верхней границе достигаемой погрешности (0,00006 нм). По-видимому, воспроизводимость наноструктуры не являлась полной, но различия были значительно меньше, чем найденные для образцов, полученных при разных потенциалах осаждения.
![]() Необходимо, однако, отметить, что полной однородности осадка вдоль поверхности при получении образцов значительной площади гарантировать нельзя. Очевидно, что такая неоднородность должна быть тем более выражена, чем больше поверхность подложки, причем эти осложняющие обстоятельства в большей степени проявляются при более высоких токах (меньших Еd), когда выше омическое падение потенциала в пространстве между различными участками поверхности и кончиком капилляра Луггина.
Необходимо, однако, отметить, что полной однородности осадка вдоль поверхности при получении образцов значительной площади гарантировать нельзя. Очевидно, что такая неоднородность должна быть тем более выражена, чем больше поверхность подложки, причем эти осложняющие обстоятельства в большей степени проявляются при более высоких токах (меньших Еd), когда выше омическое падение потенциала в пространстве между различными участками поверхности и кончиком капилляра Луггина.
![]() Количество нанесенного палладия составляло около 1 мг/см2 видимой поверхности подложки. Массу осадка определяли после промывки и высушивания, повторяя взвешивание независимо не менее 5 раз. Затем проводили тестовые измерения равновесных кривых заряжения в растворе 0.5 М Н2SO4 для того, чтобы подтвердить для полученных образцов соответствие величин Н/Pd данным подробного исследования [12]. Многократного гидрирования-дегидрирования, которое могло вызвать структурные трансформации, избегали. Образцы извлекали из раствора и приступали к структурным исследованиям после осуществления электрохимической десорбции водорода.
Количество нанесенного палладия составляло около 1 мг/см2 видимой поверхности подложки. Массу осадка определяли после промывки и высушивания, повторяя взвешивание независимо не менее 5 раз. Затем проводили тестовые измерения равновесных кривых заряжения в растворе 0.5 М Н2SO4 для того, чтобы подтвердить для полученных образцов соответствие величин Н/Pd данным подробного исследования [12]. Многократного гидрирования-дегидрирования, которое могло вызвать структурные трансформации, избегали. Образцы извлекали из раствора и приступали к структурным исследованиям после осуществления электрохимической десорбции водорода.
![]() Рентгенографические исследования проводили на дифрактометре ДРОН –3М с использованием Cu Ka излучения с автоматической регистрацией спектров и с компьютерным управлением, а также на дифрактометре Philips DX’Pert с монокристальным графитовым монохроматором. Рентгенограммы, получаемые на ДРОН-3М, регистрировались в интервале углов 2q = 38 - 127°. В области Брэгговского рефлекса запись велась через 0,01 - 0,02° с накоплением в точке в течение 10 – 30 сек в зависимости от интенсивности пика. Ошибки в определении угла 2q составляли 0,007° для менее диффузных линий и 0,03° – 0,04° для значительно уширенных рефлексов. Разделение пиков Pt и Pd и выделение их дублетных составляющих проводилось с помощью программы ProfitVZ [19], использующей профильный анализ Брегговских рефлексов. Подгоночные операции были неудовлетворительными при разложении сложных пиков, образованных линиями 220 и 311, т. к. интенсивности Pt и Pd различались на 2 – 3 порядка, вследствие текстурированности Pt подложки. Для рефлексов с сопоставимыми интенсивностями Pt и Pd положения линий Pt использовали как внутренний стандарт (это оправдывалось крупнокристаллическим характером Pt, для которой величины ОКР на 1,5-2 порядка выше, чем для Рd). Для вычисления периода элементарной ячейки a Рd использованы рефлексы 111, 200, 331 и 420, и значение a определялось путем экстраполяции зависимости a от cos 2q/sinq к q = 90°. Для разделения вкладов дисперсности и микронапряжения (микродеформации) в уширение Брэгговского рефлекса использовались рентгеновские данные, полученные на монохроматическом излучении на дифрактометре Philips DX’Pert.
Рентгенографические исследования проводили на дифрактометре ДРОН –3М с использованием Cu Ka излучения с автоматической регистрацией спектров и с компьютерным управлением, а также на дифрактометре Philips DX’Pert с монокристальным графитовым монохроматором. Рентгенограммы, получаемые на ДРОН-3М, регистрировались в интервале углов 2q = 38 - 127°. В области Брэгговского рефлекса запись велась через 0,01 - 0,02° с накоплением в точке в течение 10 – 30 сек в зависимости от интенсивности пика. Ошибки в определении угла 2q составляли 0,007° для менее диффузных линий и 0,03° – 0,04° для значительно уширенных рефлексов. Разделение пиков Pt и Pd и выделение их дублетных составляющих проводилось с помощью программы ProfitVZ [19], использующей профильный анализ Брегговских рефлексов. Подгоночные операции были неудовлетворительными при разложении сложных пиков, образованных линиями 220 и 311, т. к. интенсивности Pt и Pd различались на 2 – 3 порядка, вследствие текстурированности Pt подложки. Для рефлексов с сопоставимыми интенсивностями Pt и Pd положения линий Pt использовали как внутренний стандарт (это оправдывалось крупнокристаллическим характером Pt, для которой величины ОКР на 1,5-2 порядка выше, чем для Рd). Для вычисления периода элементарной ячейки a Рd использованы рефлексы 111, 200, 331 и 420, и значение a определялось путем экстраполяции зависимости a от cos 2q/sinq к q = 90°. Для разделения вкладов дисперсности и микронапряжения (микродеформации) в уширение Брэгговского рефлекса использовались рентгеновские данные, полученные на монохроматическом излучении на дифрактометре Philips DX’Pert.
![]() Рентгеновские электронные спектры остовных уровней и валентной зоны регистрировали на электронном спектрометре ESCALab 5 MKII (VG Gsi) с использованием Mg Ka излучения при давлении остаточных газов в аналитической камере прибора меньше 10-8 Па. Энергетическую шкалу калибровали по положению максимума линии Cu 3d 3/2 и энергии уровня Ферми материала спектрометра – меди. Для очистки от адсорбированных примесей использовали мягкий режим бомбардировки пучком ионов аргона с энергией 1,5 кэВ и плотностью 50 мкА/см2.
Рентгеновские электронные спектры остовных уровней и валентной зоны регистрировали на электронном спектрометре ESCALab 5 MKII (VG Gsi) с использованием Mg Ka излучения при давлении остаточных газов в аналитической камере прибора меньше 10-8 Па. Энергетическую шкалу калибровали по положению максимума линии Cu 3d 3/2 и энергии уровня Ферми материала спектрометра – меди. Для очистки от адсорбированных примесей использовали мягкий режим бомбардировки пучком ионов аргона с энергией 1,5 кэВ и плотностью 50 мкА/см2.
![]() Данные просвечивающей электронной микроскопии (ПЭМ) получены на приборе JEM-100C на образцах, отделенных от наружного слоя ЭОРd. Такая процедура была возможна только для осадков с массами от 2-3 мг/см2, более тонкие осадки обладали хорошей адгезией.
Данные просвечивающей электронной микроскопии (ПЭМ) получены на приборе JEM-100C на образцах, отделенных от наружного слоя ЭОРd. Такая процедура была возможна только для осадков с массами от 2-3 мг/см2, более тонкие осадки обладали хорошей адгезией.
![]() Путем разложения по описанному выше методу перекрывающихся линий Pt и Pd определены положения максимумов, интенсивности и полуширина ( В ) рефлексов 111, 200, 222, 331 и 420 каждой фазы. Соотношения интенсивностей рефлексов близки к соответствующим соотношениям для крупно кристаллического Pd. Так в Pd26 интенсивности этих рефлексов относятся как 100:40:5:14:10 и мало отличаются от справочных данных для крупно кристаллического Pd (100:50:1:5:5) [19,20], т.е. признаки текстуры практически отсутствуют.
Путем разложения по описанному выше методу перекрывающихся линий Pt и Pd определены положения максимумов, интенсивности и полуширина ( В ) рефлексов 111, 200, 222, 331 и 420 каждой фазы. Соотношения интенсивностей рефлексов близки к соответствующим соотношениям для крупно кристаллического Pd. Так в Pd26 интенсивности этих рефлексов относятся как 100:40:5:14:10 и мало отличаются от справочных данных для крупно кристаллического Pd (100:50:1:5:5) [19,20], т.е. признаки текстуры практически отсутствуют.
![]() Рассчитанные значения а для исследуемых осадков Pd26, Pd80 и Pd120 приведены в Таблице наряду с а других н.к. и к.к. Pd материалов. Значения а для Pd120 совпадают со значениями, измеренными для ЭО Pd в [17], но оказываются ниже, чем а для Pd26, Pd80. Сопоставление наших данных с данными по периоду ячейки н.к. компактов и к.к. Pd материалов, взятые из литературы [4,10,19,20], оказывается не очень информативным, т.к. существуют некоторые различия между значениями а для одного и того же материала, измеренными разными методами, однако значения а наших образцов укладываются в этот интервал.
Рассчитанные значения а для исследуемых осадков Pd26, Pd80 и Pd120 приведены в Таблице наряду с а других н.к. и к.к. Pd материалов. Значения а для Pd120 совпадают со значениями, измеренными для ЭО Pd в [17], но оказываются ниже, чем а для Pd26, Pd80. Сопоставление наших данных с данными по периоду ячейки н.к. компактов и к.к. Pd материалов, взятые из литературы [4,10,19,20], оказывается не очень информативным, т.к. существуют некоторые различия между значениями а для одного и того же материала, измеренными разными методами, однако значения а наших образцов укладываются в этот интервал.
![]() Отражения всех исследованных осадков заметно уширены по сравнению с линиями более равновесной и крупно кристаллической Pd фольги, причем для Pd26 в большей степени, чем для Pd80 и Pd120. В случае Pd26 полуширина В для отражений 111, 200, 222, 331 и 420 составляет, соответственно, 0,460; 0,730; 1,150; 1,775 и 2,200. Уширение линий может быть вызвано участием в дифракции малых кристаллитов (размеры ОКР, т.е. размеры зерен D<100 нм), присутствием дефектов, вызывающих упругие напряжения и приводящих к микродеформациям кристаллов ( e = Dа/a0 ), а также флуктуациями в распределении микронапряжений в материале. Для микродеформаций характерна угловая зависимость величины уширения рефлексов, которая и наблюдается для всех исследованных осадков, но особенно выражена в Pd26. Уширение, вызванное малыми размерами частиц, не зависит от брэгговского угла. Поэтому обычно для разделения обоих вкладов в уширение рефлексов (и определения размеров ОКР и микродеформаций) анализируются профили пиков от одного и того же семейства плоскостей, но с разным порядком отражения. Из-за методических трудностей, описанных выше, оказалось невозможным точно определить профили таких семейств плоскостей, поэтому расчет размеров ОКР и микродеформаций проводили по модифицированнному методу [21] из анализа профиля одного максимума для рефлексов 111, 200 и 420 (Таблица).
Отражения всех исследованных осадков заметно уширены по сравнению с линиями более равновесной и крупно кристаллической Pd фольги, причем для Pd26 в большей степени, чем для Pd80 и Pd120. В случае Pd26 полуширина В для отражений 111, 200, 222, 331 и 420 составляет, соответственно, 0,460; 0,730; 1,150; 1,775 и 2,200. Уширение линий может быть вызвано участием в дифракции малых кристаллитов (размеры ОКР, т.е. размеры зерен D<100 нм), присутствием дефектов, вызывающих упругие напряжения и приводящих к микродеформациям кристаллов ( e = Dа/a0 ), а также флуктуациями в распределении микронапряжений в материале. Для микродеформаций характерна угловая зависимость величины уширения рефлексов, которая и наблюдается для всех исследованных осадков, но особенно выражена в Pd26. Уширение, вызванное малыми размерами частиц, не зависит от брэгговского угла. Поэтому обычно для разделения обоих вкладов в уширение рефлексов (и определения размеров ОКР и микродеформаций) анализируются профили пиков от одного и того же семейства плоскостей, но с разным порядком отражения. Из-за методических трудностей, описанных выше, оказалось невозможным точно определить профили таких семейств плоскостей, поэтому расчет размеров ОКР и микродеформаций проводили по модифицированнному методу [21] из анализа профиля одного максимума для рефлексов 111, 200 и 420 (Таблица).
![]() Принималось, что профиль рентгеновской дифракционной линии описывается функцией Фойгта, которая является комбинацией функции Коши и Гаусса. Экспериментально измеренный профиль (h) является комбинацией структурно уширенного профиля (f) и стандартного профиля (g). Стандартный профиль используется для учета инструментального уширения. Тогда
Принималось, что профиль рентгеновской дифракционной линии описывается функцией Фойгта, которая является комбинацией функции Коши и Гаусса. Экспериментально измеренный профиль (h) является комбинацией структурно уширенного профиля (f) и стандартного профиля (g). Стандартный профиль используется для учета инструментального уширения. Тогда
| hc = gc * fc и hG = gG * fG, |
где индексы C и G означают компоненты Коши и Гаусса профиля Фойгта, а звездочка (*) - символ операции свертки. Отсюда следует, что интегральные ширины для профиля f (bfC и bfG) после коррекции инструментального уширения даются выражениями:
| bfC = bhС – bGC и (bfG)2 = (bhG)2 – (bGG)2. |
Составляющие Коши и Гаусса могут быть получены из b и отношения 2w/b для h и g профилей, где 2w – ширина на половине высоты рентгеновского пика и b – интегральная ширина. Были получены следующие эмпирические уравнения:
| bС = b[2.0207 – 0.4803(2w/b) – 1.7756(2w/b)2] |
| bG = b{0.6420 + 1.4187[(2w/b) – 2/p]1/2 – 2.2043(2w/b) + 1.8706(2w/b)2}. |
Размеры ОКР (D) и микродеформации (e) могут быть получены из выражений:
| D = K g/ bfC cos q, e = bfG/ 4 tg q, |
где коэффициент К = 0.9.
![]() Анализ структурных параметров Рd26, Рd80 и Рd120, приведенных в Таблице, показывает, что существует корреляция структурных параметров и потенциала осаждения Еd: чем выше перенапряжение, тем выше период элементарной ячейки и микродеформация (микронапряжения) и тем меньше размер кристаллита (или ОКР). Далее, сопоставление величин деформации, данных в Таблице, демонстрирует, что в Рd26, Рd80 они близки к значениям в н.к. Рd и существенно выше, чем в к.к. Рd. Кроме того, если в последнем микродеформации изотропны, то в н.к. Рd и в ЭОРd они анизотропны.
Анализ структурных параметров Рd26, Рd80 и Рd120, приведенных в Таблице, показывает, что существует корреляция структурных параметров и потенциала осаждения Еd: чем выше перенапряжение, тем выше период элементарной ячейки и микродеформация (микронапряжения) и тем меньше размер кристаллита (или ОКР). Далее, сопоставление величин деформации, данных в Таблице, демонстрирует, что в Рd26, Рd80 они близки к значениям в н.к. Рd и существенно выше, чем в к.к. Рd. Кроме того, если в последнем микродеформации изотропны, то в н.к. Рd и в ЭОРd они анизотропны.
![]() Меньшие значения а у образцов Рd80 и Рd120 по сравнению с а массивного крупнокристаллического Pd (0,38902 нм) можно, как и в работе [3], отнести за счет микронапряжений, вызванных дефектами разупорядочения и появляющихся в кристаллитах в неравновесных условиях кристаллизации, а также присутствием вакансий, остающихся в Pd решетке после удаления атомов Н из образующегося в ходе роста осадка гидрида Pd. Однако большие (по сравнению с образцами Рd80 и Рd120) микронапряжения в образце Рd26, обладающем периодом а, равным а массивного Pd, показывают, что наряду с напряжением на строение кристаллической фазы образца Рd26 влияют и друие факторы.
Меньшие значения а у образцов Рd80 и Рd120 по сравнению с а массивного крупнокристаллического Pd (0,38902 нм) можно, как и в работе [3], отнести за счет микронапряжений, вызванных дефектами разупорядочения и появляющихся в кристаллитах в неравновесных условиях кристаллизации, а также присутствием вакансий, остающихся в Pd решетке после удаления атомов Н из образующегося в ходе роста осадка гидрида Pd. Однако большие (по сравнению с образцами Рd80 и Рd120) микронапряжения в образце Рd26, обладающем периодом а, равным а массивного Pd, показывают, что наряду с напряжением на строение кристаллической фазы образца Рd26 влияют и друие факторы.
![]() Была предпринята попытка дополнительно охарактеризовать различия дефектности исследуемых образцов Pd26 и Pd120 по наблюдениям эволюции их структурных параметров при температурной обработке в интервале температур от 20 до 400 – 450°C. После отжига на воздухе образцы охлаждали на массивной подложке и производили измерения при комнатной температуре. Из рис. 1 видно, что период а образца Рd120 практически линейно увеличивается с увеличением температуры отжига, достигая значений а объемного Pd (кривая 2). Аналогичные результаты приведены в [22] для электролитических осадков платины, отделенных от подложки, в более позднем рентенографическом исследовании [18] аналогичных опытов не проводили. Существенно, что с увеличением температуры отжига происходило уменьшение уширения рефлексов при неизменной их интегральной интенсивности. Величина В рефлекса 331 уменьшилась на 27%, причем изменение положения максимума рефлекса (0,13°) при увеличении температуры от 20 до 450°C оказалось равным половине изменения В (0,125°). Так как температура отжига была существенно ниже температуры рекристаллизации (равной примерно 0,4 Тпл ), при которой можно было бы ожидать изменения размера кристаллитов, изменившийся вклад в уширение очевидно следует отнести за счет уменьшения микронапряжений в кристаллической решетке Рd120. Отжиг устраняет дефекты (возможно, вакансии решетки Pd) и связанные с ними микронапряжения и микродеформацию ячейки. Это означает, что в образце Рd120 только микронапряжения обуславливают заниженную по сравнению с массивным Pd величину периода элементарной ячейки. Оценка e по изменению величины периода ячейки (0,0005 нм) дает Dа/а0 = 1,3*10-3, что хорошо согласуется со средним значением e, полученным из анализа профиля пиков.
Была предпринята попытка дополнительно охарактеризовать различия дефектности исследуемых образцов Pd26 и Pd120 по наблюдениям эволюции их структурных параметров при температурной обработке в интервале температур от 20 до 400 – 450°C. После отжига на воздухе образцы охлаждали на массивной подложке и производили измерения при комнатной температуре. Из рис. 1 видно, что период а образца Рd120 практически линейно увеличивается с увеличением температуры отжига, достигая значений а объемного Pd (кривая 2). Аналогичные результаты приведены в [22] для электролитических осадков платины, отделенных от подложки, в более позднем рентенографическом исследовании [18] аналогичных опытов не проводили. Существенно, что с увеличением температуры отжига происходило уменьшение уширения рефлексов при неизменной их интегральной интенсивности. Величина В рефлекса 331 уменьшилась на 27%, причем изменение положения максимума рефлекса (0,13°) при увеличении температуры от 20 до 450°C оказалось равным половине изменения В (0,125°). Так как температура отжига была существенно ниже температуры рекристаллизации (равной примерно 0,4 Тпл ), при которой можно было бы ожидать изменения размера кристаллитов, изменившийся вклад в уширение очевидно следует отнести за счет уменьшения микронапряжений в кристаллической решетке Рd120. Отжиг устраняет дефекты (возможно, вакансии решетки Pd) и связанные с ними микронапряжения и микродеформацию ячейки. Это означает, что в образце Рd120 только микронапряжения обуславливают заниженную по сравнению с массивным Pd величину периода элементарной ячейки. Оценка e по изменению величины периода ячейки (0,0005 нм) дает Dа/а0 = 1,3*10-3, что хорошо согласуется со средним значением e, полученным из анализа профиля пиков.
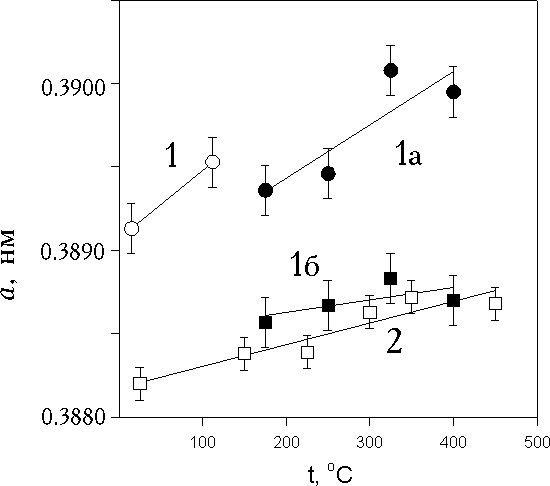 |
| Рис. 1. Периоды элементарной ячейки палладия в образцах Pd26 и Pd120 в зависимости от температуры отжига: 1, 1а и 1б - Pd26, 2 - Pd120 |
![]() Поведение образца Рd26 при отжиге оказалось более сложным (кривые 1, 1а и 1б рис.1). Образец, рентгенограмма которого до термообработки соответствовала однофазному осадку, в интервале температур 112 – 175°C претерпевает разложение на две фазы с такими же кубическими решетками, что и Pd. Параметр а одной из фаз ( с меньшими значениями а ) с изменением Т отжига ведет себя как в образце Рd120 (кривые 1б и 2 рис.1), а период второй фазы более существенно увеличивается при возрастании Т вплоть до 400°C (кривая 1а рис. 1). Кроме того, суммарные интенсивности пиков обеих фаз уменьшаются примерно на 30% по сравнению с исходной интенсивностью пика в неотожженном Рd26. Эти результаты можно интерпретировать как следствие сосуществования областей с дефектностью того же типа, что и в образце Рd120, и областей с дефектностью совершенно иного типа.
Поведение образца Рd26 при отжиге оказалось более сложным (кривые 1, 1а и 1б рис.1). Образец, рентгенограмма которого до термообработки соответствовала однофазному осадку, в интервале температур 112 – 175°C претерпевает разложение на две фазы с такими же кубическими решетками, что и Pd. Параметр а одной из фаз ( с меньшими значениями а ) с изменением Т отжига ведет себя как в образце Рd120 (кривые 1б и 2 рис.1), а период второй фазы более существенно увеличивается при возрастании Т вплоть до 400°C (кривая 1а рис. 1). Кроме того, суммарные интенсивности пиков обеих фаз уменьшаются примерно на 30% по сравнению с исходной интенсивностью пика в неотожженном Рd26. Эти результаты можно интерпретировать как следствие сосуществования областей с дефектностью того же типа, что и в образце Рd120, и областей с дефектностью совершенно иного типа.
![]() “Расслоение” ЭОРd26 после достаточно слабого нагрева не исключает предположения о гетерогенности исходного материала, несмотря на отсутствие ее прямых рентгенографических проявлений. Увеличение параметра решетки в значительной части объема ЭОРd26 (кривая 1а, рис. 1 ) и высокое по сравнению с другими образцами значение этого параметра в исходном материале (при наличии самых высоких микродеформаций) принципиально отличает обсуждаемый осадок от аналогов, полученных при других потенциалах осаждения – для них характерно значимое снижение параметра по сравнению с равновесной величиной ([17,18], данные настоящей работы для Рd80 и Рd120).
“Расслоение” ЭОРd26 после достаточно слабого нагрева не исключает предположения о гетерогенности исходного материала, несмотря на отсутствие ее прямых рентгенографических проявлений. Увеличение параметра решетки в значительной части объема ЭОРd26 (кривая 1а, рис. 1 ) и высокое по сравнению с другими образцами значение этого параметра в исходном материале (при наличии самых высоких микродеформаций) принципиально отличает обсуждаемый осадок от аналогов, полученных при других потенциалах осаждения – для них характерно значимое снижение параметра по сравнению с равновесной величиной ([17,18], данные настоящей работы для Рd80 и Рd120).
![]() Высокое значение а можно было бы связывать, вероятно, с внедрением каких-либо посторонних атомов. В качестве гипотезы можно предложить также остаточную деформацию решетки палладиевой матрицы, возникшую в ходе гидрирования осадка непосредственно при формировании последнего: ведь именно сопутствующее формирование b-фазы гидрида специфично для Рd26 и не имело места для других образцов. Внедрение в процессе приготовления других легких атомов, приводящее к увеличению параметра решетки наноразмерного палладия (как, например, в [23]), представляется менее вероятным, поскольку все исследованные осадки получены в одинаковой среде. Сплавообразование с платиной следует отвергнуть, т.к. ни период ячейки, ни интенсивность рентгеновских отражений платины после отжига Рd26 не изменяются. Углерод, обнаруживаемый на поверхности образца с помощью РЭС в количестве 3 – 5 ат.% , находится в виде углеводородных и карбоксосодержащих соединений и их пиролитического остатка, но не в форме карбида, поэтому внедрение углерода можно надежно исключить.
Высокое значение а можно было бы связывать, вероятно, с внедрением каких-либо посторонних атомов. В качестве гипотезы можно предложить также остаточную деформацию решетки палладиевой матрицы, возникшую в ходе гидрирования осадка непосредственно при формировании последнего: ведь именно сопутствующее формирование b-фазы гидрида специфично для Рd26 и не имело места для других образцов. Внедрение в процессе приготовления других легких атомов, приводящее к увеличению параметра решетки наноразмерного палладия (как, например, в [23]), представляется менее вероятным, поскольку все исследованные осадки получены в одинаковой среде. Сплавообразование с платиной следует отвергнуть, т.к. ни период ячейки, ни интенсивность рентгеновских отражений платины после отжига Рd26 не изменяются. Углерод, обнаруживаемый на поверхности образца с помощью РЭС в количестве 3 – 5 ат.% , находится в виде углеводородных и карбоксосодержащих соединений и их пиролитического остатка, но не в форме карбида, поэтому внедрение углерода можно надежно исключить.
![]() Таким образом, наиболее вероятной причиной аномального поведения структурных параметров Рd26 следует считать влияние остаточной деформации на некоторую часть кристаллитов, подвергшихся значительной дилатации при десорбции водорода ( при фазовом b <=> a переходе происходит 10% уменьшение объема кристаллической решетки). Данная ситуация аналогична распаду пересыщенного твердого раствора, при котором выделяющаяся из твердого раствора фаза имеет решетку, отличающуюся от матрицы сплава и другой удельный объем; на границах областей выделившейся фазы и матрицы создаются микроискажения, сохраняющиеся из-за наличия сопряжения ( когерентности) решеток [24]. Если кристаллиты обеих фаз равномерно распределены по объему материала и имеют общие межзеренные границы, то нельзя исключить ситуацию, при которой релаксация одной из них приводит к увеличению дефектности другой.
Таким образом, наиболее вероятной причиной аномального поведения структурных параметров Рd26 следует считать влияние остаточной деформации на некоторую часть кристаллитов, подвергшихся значительной дилатации при десорбции водорода ( при фазовом b <=> a переходе происходит 10% уменьшение объема кристаллической решетки). Данная ситуация аналогична распаду пересыщенного твердого раствора, при котором выделяющаяся из твердого раствора фаза имеет решетку, отличающуюся от матрицы сплава и другой удельный объем; на границах областей выделившейся фазы и матрицы создаются микроискажения, сохраняющиеся из-за наличия сопряжения ( когерентности) решеток [24]. Если кристаллиты обеих фаз равномерно распределены по объему материала и имеют общие межзеренные границы, то нельзя исключить ситуацию, при которой релаксация одной из них приводит к увеличению дефектности другой.
![]() На основе различного поведения кристаллических решеток Рd26 и Рd120 при отжиге (в Рd26 уменьшались и интенсивность, и уширение линий обеих фаз – Рd -а и Рd -б, а в Рd120 произошло лишь уменьшение уширения рефлексов) можно сделать некоторые предположения о преобладающих дефектах в этих осадках в терминах представлений о дефектах первого и второго классов [25]. Дефекты первого класса, для которых наблюдается ослабление интенсивности рентгеновских рефлексов, характеризуются малым ареалом действия, и возмущения в них (смещения атомов) убывают с расстоянием как 1/r2. Дефекты второго класса – протяженные, и создаваемые ими смещения убывают с расстоянием более медленно, как 1/ r2/3. К таким дефектам относятся линейные, краевые дислокации, при концентрации которых выше 1010 см наблюдаются уширение рефлексов. Если принять во внимание размер нанокристаллитов в Рd26 – 6,5 нм, наличие в них более ограниченных дефектов первого класса вполне объяснимо. В кристаллитах Рd120 с большим размером зерен, очевидно, становится возможным и образование более протяженных дефектов – дислокаций. Экспериментально подтвердить наличие дислокаций разных типов в нанокристаллитах меди большего размера (около 20 нм) удалось лишь недавно с помощью специальной рентгеновской техники высокого разрешения [7].
На основе различного поведения кристаллических решеток Рd26 и Рd120 при отжиге (в Рd26 уменьшались и интенсивность, и уширение линий обеих фаз – Рd -а и Рd -б, а в Рd120 произошло лишь уменьшение уширения рефлексов) можно сделать некоторые предположения о преобладающих дефектах в этих осадках в терминах представлений о дефектах первого и второго классов [25]. Дефекты первого класса, для которых наблюдается ослабление интенсивности рентгеновских рефлексов, характеризуются малым ареалом действия, и возмущения в них (смещения атомов) убывают с расстоянием как 1/r2. Дефекты второго класса – протяженные, и создаваемые ими смещения убывают с расстоянием более медленно, как 1/ r2/3. К таким дефектам относятся линейные, краевые дислокации, при концентрации которых выше 1010 см наблюдаются уширение рефлексов. Если принять во внимание размер нанокристаллитов в Рd26 – 6,5 нм, наличие в них более ограниченных дефектов первого класса вполне объяснимо. В кристаллитах Рd120 с большим размером зерен, очевидно, становится возможным и образование более протяженных дефектов – дислокаций. Экспериментально подтвердить наличие дислокаций разных типов в нанокристаллитах меди большего размера (около 20 нм) удалось лишь недавно с помощью специальной рентгеновской техники высокого разрешения [7].
![]() Рентгенофотоэлектронные спектры остовного уровня Pd 3d исследуемых образцов представлены на рис.2, 3. Спектр образца Рd26 состоит из одиночного дублета Pd 3d 3/2 (Есв = 335,0 эВ) и Pd 3d 5/2 (Есв= 340,3 эВ). Для чистой поверхности Pd характерна несколько большая Есв (335,7 и 341,0, соответственно).
Рентгенофотоэлектронные спектры остовного уровня Pd 3d исследуемых образцов представлены на рис.2, 3. Спектр образца Рd26 состоит из одиночного дублета Pd 3d 3/2 (Есв = 335,0 эВ) и Pd 3d 5/2 (Есв= 340,3 эВ). Для чистой поверхности Pd характерна несколько большая Есв (335,7 и 341,0, соответственно).
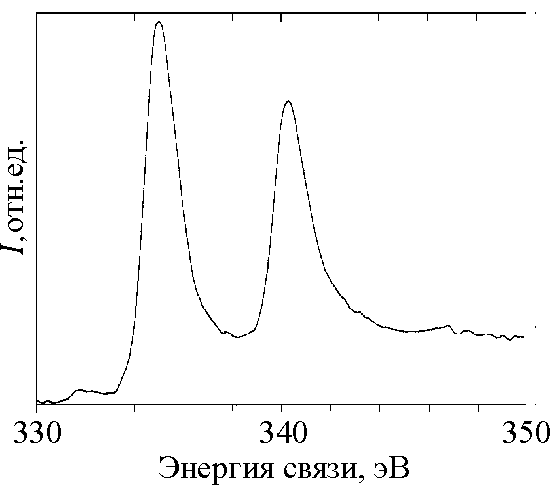 |
| Рис. 2 Ренгеноэлектронный спектр остовного уровня Pd 3d образца Pd26. |
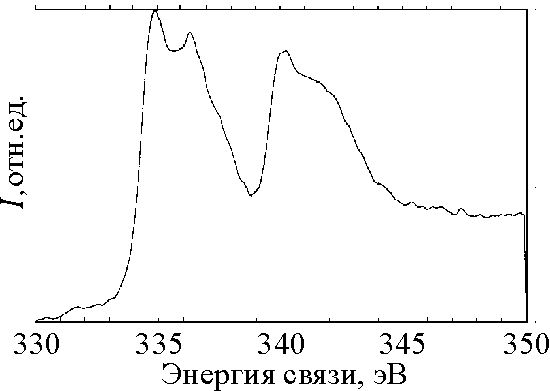 |
| Рис. 3 Ренгеноэлектронный спектр остовного уровня Pd 3d образца Pd120. |
![]() Спектр 3d Pd в образце Рd26 обладает рядом особенностей, которые отличают его от спектра чистой поверхности к.к.Pd:
Спектр 3d Pd в образце Рd26 обладает рядом особенностей, которые отличают его от спектра чистой поверхности к.к.Pd:
![]() “Shake up” сателлиты и асимметрию в случае Pd объясняют обычно наличием незаполненных d – состояний, расположенных непосредственно над уровнем Ферми. Соответственно, отсутствие указанных проявлений свидетельствует о заполнении этих состояний. Спектр 3d Pd образца Рd26 обнаруживает сходство с аналогичным спектром гидрида PdHx [26]. Для последнего, однако, В' меньше (1.2 эВ), чем для чистого Pd (1.4 эВ).
“Shake up” сателлиты и асимметрию в случае Pd объясняют обычно наличием незаполненных d – состояний, расположенных непосредственно над уровнем Ферми. Соответственно, отсутствие указанных проявлений свидетельствует о заполнении этих состояний. Спектр 3d Pd образца Рd26 обнаруживает сходство с аналогичным спектром гидрида PdHx [26]. Для последнего, однако, В' меньше (1.2 эВ), чем для чистого Pd (1.4 эВ).
![]() Спектры образцов Рd80 и Рd120 демонстрируют сдвоенные дублеты: энергия связи первого пика такая же, как для Рd26, а второй пик обладает более высокой энергией связи (336,3 эВ). Пики 3d Pd с большей нежели для чистого Pd, но меньшей, чем для фазы PdО-(ОН) х энергией связи (Есв = 339 эВ) [27], обнаруживали и ранее на поверхности Рd. Такие эффекты давал не монослой адсорбированного кислорода, а тонкие (нано) пленки состава Pd-Ох [28]. Действительно, на поверхности образцов Рd80 и Рd120, в отличие от Рd26, после мягкой очистки ионами аргона наблюдается значительное количество кислорода (по данным рентгеноспектрального микроанализа – до 5 ат%). Картины спектров для Рd80 и Рd120, получаемых с углами выхода электронов d =20° и 60° (глубина выхода электронов 5-10А и 20-25А, соответственно) различались. В спектрах, получаемых от “объемного” слоя (d =60°) уменьшался вклад Pd-Ох и увеличивался вклад пиков от чистого Рd. Это означает, что, в отличие от образца Рd26, поверхностный слой (толщиной ~ 2 нм) образцов Рd80 и Рd120 существенно окислен.
Спектры образцов Рd80 и Рd120 демонстрируют сдвоенные дублеты: энергия связи первого пика такая же, как для Рd26, а второй пик обладает более высокой энергией связи (336,3 эВ). Пики 3d Pd с большей нежели для чистого Pd, но меньшей, чем для фазы PdО-(ОН) х энергией связи (Есв = 339 эВ) [27], обнаруживали и ранее на поверхности Рd. Такие эффекты давал не монослой адсорбированного кислорода, а тонкие (нано) пленки состава Pd-Ох [28]. Действительно, на поверхности образцов Рd80 и Рd120, в отличие от Рd26, после мягкой очистки ионами аргона наблюдается значительное количество кислорода (по данным рентгеноспектрального микроанализа – до 5 ат%). Картины спектров для Рd80 и Рd120, получаемых с углами выхода электронов d =20° и 60° (глубина выхода электронов 5-10А и 20-25А, соответственно) различались. В спектрах, получаемых от “объемного” слоя (d =60°) уменьшался вклад Pd-Ох и увеличивался вклад пиков от чистого Рd. Это означает, что, в отличие от образца Рd26, поверхностный слой (толщиной ~ 2 нм) образцов Рd80 и Рd120 существенно окислен.
![]() Поскольку процесс электроосаждения и предварительные эксперименты по сорбции-десорбции водорода выполнялись в условиях отсутствия на поверхности металла адсорбированного кислорода, следует предположить, что окисление поверхности происходило на воздухе после завершения всех электрохимических экспериментов. Более высокая устойчивость Рd26 к окислению не находит пока однозначного объяснения. На первый взгляд, этот результат противоречит описанной в [12] тенденции к уменьшению затрат заряда на адсорбцию кислорода с ростом Еd для исследуемой группы осадков (в [12] – ЭОРdI). Однако, как было показано в [18], отнесение количества адсорбированного кислорода к истинной поверхности, определенной по адсорбции меди, не является вполне надежным из-за высокой чувствительности заполнений по меди к особенностям структуры поверхности осадков. Ниже обоснованы некоторые предположения о природе найденного различия.
Поскольку процесс электроосаждения и предварительные эксперименты по сорбции-десорбции водорода выполнялись в условиях отсутствия на поверхности металла адсорбированного кислорода, следует предположить, что окисление поверхности происходило на воздухе после завершения всех электрохимических экспериментов. Более высокая устойчивость Рd26 к окислению не находит пока однозначного объяснения. На первый взгляд, этот результат противоречит описанной в [12] тенденции к уменьшению затрат заряда на адсорбцию кислорода с ростом Еd для исследуемой группы осадков (в [12] – ЭОРdI). Однако, как было показано в [18], отнесение количества адсорбированного кислорода к истинной поверхности, определенной по адсорбции меди, не является вполне надежным из-за высокой чувствительности заполнений по меди к особенностям структуры поверхности осадков. Ниже обоснованы некоторые предположения о природе найденного различия.
![]() На рис.4а представлены спектры валентных зон Рd26, к.к. Pd и образца Рd120 после снятия кислород-содержащего слоя с помощью длительной ионной бомбардировки. Спектр валентной зоны (В.З.) Рd26 существенно отличается от спектра В.З. чистого Pd и оказывается схожим со спектром гидрида PdHx (х»0,8), полученным в [26] (рис.4б).
На рис.4а представлены спектры валентных зон Рd26, к.к. Pd и образца Рd120 после снятия кислород-содержащего слоя с помощью длительной ионной бомбардировки. Спектр валентной зоны (В.З.) Рd26 существенно отличается от спектра В.З. чистого Pd и оказывается схожим со спектром гидрида PdHx (х»0,8), полученным в [26] (рис.4б).
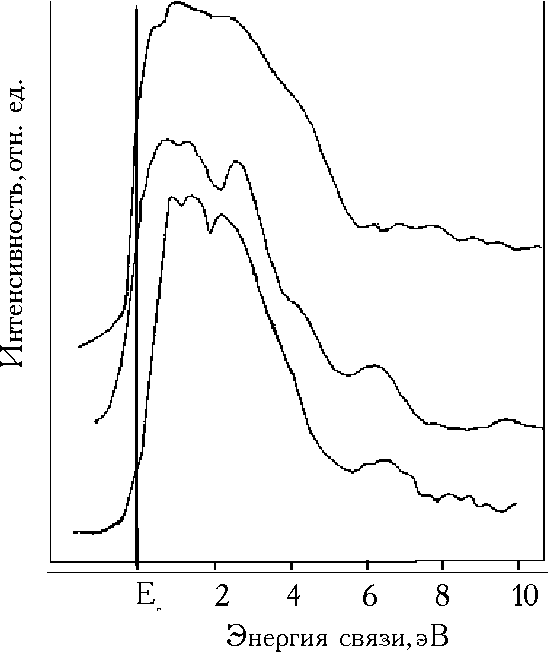 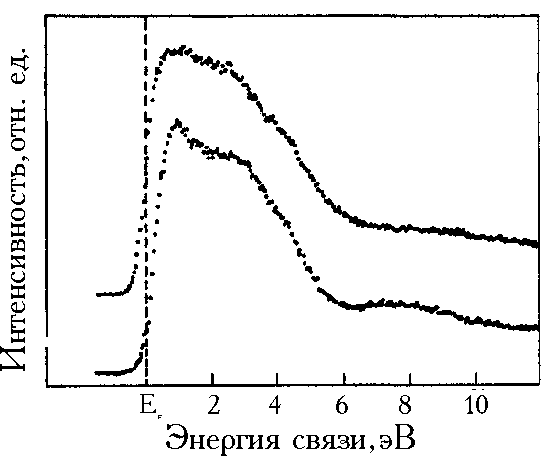 |
| Рис. 4 Спектры валентной зоны: а - образца Pd26, образца Pd120 после бомбардировки ионами аргона и фольги к.к. Pd (снизу вверх), б - PdHx (x = 0,8) (снизу) и монокристалла Pd (сверху) [26]. |
![]() Гидриды Pd представляют собой пример систем, в которых образующиеся гибридные связи Pd–H приводят как к заполнению пустых d валентных состояний (и, соответственно, к устранению эффектов, связанных с shake-up электронами), так и к формированию новых Pd–H состояний, находящихся в области 8 эВ в В.З. PdHx. Это показано целым рядом экспериментальных работ и расчетами зонной структуры PdHx (см., например, в [26]). Реорганизация электронной структуры PdHx в сравнении с к.к. Pd проявляется в уменьшении плотности состояний (сужение пика 4d– электронов Pd) и небольшом (0,2 – 0,3 эВ) понижении уровня Ферми металла.
Гидриды Pd представляют собой пример систем, в которых образующиеся гибридные связи Pd–H приводят как к заполнению пустых d валентных состояний (и, соответственно, к устранению эффектов, связанных с shake-up электронами), так и к формированию новых Pd–H состояний, находящихся в области 8 эВ в В.З. PdHx. Это показано целым рядом экспериментальных работ и расчетами зонной структуры PdHx (см., например, в [26]). Реорганизация электронной структуры PdHx в сравнении с к.к. Pd проявляется в уменьшении плотности состояний (сужение пика 4d– электронов Pd) и небольшом (0,2 – 0,3 эВ) понижении уровня Ферми металла.
![]() Вместе с тем аналогичный эффект сужения В.З. и уменьшения плотности состояний в прифермиевской области дают сильно разупорядоченные, дефектные (в частности созданные бомбардировкой ионами Ar) поверхности Pd. Это демонстрирует спектр ВЗ, полученный в результате длительного травления ионами Ar поверхности образца ЭОРd120, который был полностью освобожден от кислорода в результате этой обработки.Этот спектр отличается от спектра В.З. PdHx наличием дополнительного пика при 6 эВ, а не при 8 эВ, как в PdHx.
Вместе с тем аналогичный эффект сужения В.З. и уменьшения плотности состояний в прифермиевской области дают сильно разупорядоченные, дефектные (в частности созданные бомбардировкой ионами Ar) поверхности Pd. Это демонстрирует спектр ВЗ, полученный в результате длительного травления ионами Ar поверхности образца ЭОРd120, который был полностью освобожден от кислорода в результате этой обработки.Этот спектр отличается от спектра В.З. PdHx наличием дополнительного пика при 6 эВ, а не при 8 эВ, как в PdHx.
![]() Сопоставление спектра ВЗ образца ЭОРd26 со спектрами PdHx, дефектной поверхности образца ЭОРd120 (после ионного травления) и чистой поверхности к.к. Pd (рис. 4а,б) показывает, что в данном случае проявляются особенности В.З. гидрида Pd и дефектной поверхности Pd и существенные отличия от поведения чистого палладия. Специально следует отметить новую полосу состояний, находящуюся в спектре образца ЭОРd26 при 6 эВ, так же, как и в образце ЭОРd120 после травления (а не при 8 эВ, как в случае PdHx). Поэтому необычную для чистого Pd картину В.З., а также спектров 3d Pd образца Рd26 можно приписать как сильной дефектности этого осадка, так и наличию остаточного водорода. Полностью исключить наличие остаточного водорода в исследуемых материалах (и, в особенности, в ЭОРd26) нельзя. Известны примеры чрезвычайно медленного транспорта водорода, сорбированного в дефектных областях [29,30]: характерные времена дегазации могут при этом на несколько порядков превышать обычные времена десорбции водорода из октаэдрических позиций в упорядоченной палладиевой матрице. В то же время крайне маловероятно, что после экспериментально наблюдавшейся в предварительных электрохимических тестах десорбции значительного количества водорода (для Рd26 Н/Pd>1) остаточная концентрация водорода в этом образце сравнима с концентрацией в b-фазе “обычного” палладия (спектры в [26] приведены именно для b-фазы гидрида). Более реалистичным представляется локализация остаточного водорода в некоторых структурно-специфичных ловушках, в качестве которых могут, в частности, выступать дефектные фрагменты приповерхностных слоев палладия. Такое предположение могло бы, в частности, непротиворечиво объяснить необычную устойчивость Рd26 к окислению на воздухе при наличии выраженной дефектности.
Сопоставление спектра ВЗ образца ЭОРd26 со спектрами PdHx, дефектной поверхности образца ЭОРd120 (после ионного травления) и чистой поверхности к.к. Pd (рис. 4а,б) показывает, что в данном случае проявляются особенности В.З. гидрида Pd и дефектной поверхности Pd и существенные отличия от поведения чистого палладия. Специально следует отметить новую полосу состояний, находящуюся в спектре образца ЭОРd26 при 6 эВ, так же, как и в образце ЭОРd120 после травления (а не при 8 эВ, как в случае PdHx). Поэтому необычную для чистого Pd картину В.З., а также спектров 3d Pd образца Рd26 можно приписать как сильной дефектности этого осадка, так и наличию остаточного водорода. Полностью исключить наличие остаточного водорода в исследуемых материалах (и, в особенности, в ЭОРd26) нельзя. Известны примеры чрезвычайно медленного транспорта водорода, сорбированного в дефектных областях [29,30]: характерные времена дегазации могут при этом на несколько порядков превышать обычные времена десорбции водорода из октаэдрических позиций в упорядоченной палладиевой матрице. В то же время крайне маловероятно, что после экспериментально наблюдавшейся в предварительных электрохимических тестах десорбции значительного количества водорода (для Рd26 Н/Pd>1) остаточная концентрация водорода в этом образце сравнима с концентрацией в b-фазе “обычного” палладия (спектры в [26] приведены именно для b-фазы гидрида). Более реалистичным представляется локализация остаточного водорода в некоторых структурно-специфичных ловушках, в качестве которых могут, в частности, выступать дефектные фрагменты приповерхностных слоев палладия. Такое предположение могло бы, в частности, непротиворечиво объяснить необычную устойчивость Рd26 к окислению на воздухе при наличии выраженной дефектности.
![]() Можно предположить, что в образце Рd26 происходит локализация части электронов проводимости на дефектах поверхностного слоя, захвативших остаточный водород, и, как следствие, имеет место уменьшение плотности электронных состояний в прифермиевской области. Большее уширение остовных уровней по сравнению с Pd и с PdHx , характерное для разупорядоченных поверхностей, поддерживает это предположение.
Можно предположить, что в образце Рd26 происходит локализация части электронов проводимости на дефектах поверхностного слоя, захвативших остаточный водород, и, как следствие, имеет место уменьшение плотности электронных состояний в прифермиевской области. Большее уширение остовных уровней по сравнению с Pd и с PdHx , характерное для разупорядоченных поверхностей, поддерживает это предположение.
![]() Размеры ОКР, установленные в настоящей работе, оказались существенно ниже, чем аналогичные в [17]. Они соответствуют области малых размеров в широких размерных распределениях, построенных для различных Рd в [12] по данным сканирующей туннельной микроскопии. Последний метод, безусловно, обладает недостатками: визуализирует только частицы, непосредственно примыкающие к наружной поверхности осадка, и не всегда позволяет различить границы сросшихся или близко расположенных частиц.
Размеры ОКР, установленные в настоящей работе, оказались существенно ниже, чем аналогичные в [17]. Они соответствуют области малых размеров в широких размерных распределениях, построенных для различных Рd в [12] по данным сканирующей туннельной микроскопии. Последний метод, безусловно, обладает недостатками: визуализирует только частицы, непосредственно примыкающие к наружной поверхности осадка, и не всегда позволяет различить границы сросшихся или близко расположенных частиц.
![]() Независимая электронно-микроскопическая характеристика ЭОРd методом просвечивающей электронной микроскопии оказалась чрезвычайно затруднена компактностью покрытий, от которых удавалось отделить только многослойные пакеты. Визуализация отдельных частиц оказывалась возможной лишь на краях пакетов (рис.5б), причем обеспечить достаточно высокое разрешение не удавалось из-за неустойчивости фрагментов осадка к воздействию электронного пучка: через несколько минут происходило сглаживание краевых фрагментов (рис. 5в), что указывает на пониженную теплопроводность ЭОРd. Столь нетипичное для металлов поведение косвенно согласуется с заключением о высокой дефектности.
Независимая электронно-микроскопическая характеристика ЭОРd методом просвечивающей электронной микроскопии оказалась чрезвычайно затруднена компактностью покрытий, от которых удавалось отделить только многослойные пакеты. Визуализация отдельных частиц оказывалась возможной лишь на краях пакетов (рис.5б), причем обеспечить достаточно высокое разрешение не удавалось из-за неустойчивости фрагментов осадка к воздействию электронного пучка: через несколько минут происходило сглаживание краевых фрагментов (рис. 5в), что указывает на пониженную теплопроводность ЭОРd. Столь нетипичное для металлов поведение косвенно согласуется с заключением о высокой дефектности.
 |
| Рис.5. Изображения наружных слоев Рd26, полученные методом просвечивающей электронной микроскопии: а, б – в начале измерений; в – после нескольких минут измерений. |
![]() По имеющимся изображениям неоплавленных фрагментов (нижняя часть рис. 5б) можно оценить верхнюю границу размера частиц в ЭОРd26: около 15 нм. Независимые опыты проводились с образцами Рd26 большой толщины, для которых рентгенографические исследования затруднялись слабой адгезией наружных слоев осадка. Для отделения тонких фрагментов слабая адгезия оказалась, напротив, благоприятна, и удалось визуализировать достаточно большое число частиц с признаками огранки и размерами 7.5 – 9.5 нм. Разумеется, строение наружных слоев осадков толщиной несколько мкм может отличаться от строения прилегающих к подложке и промежуточных слоев компактных осадков, однако полученные микроскопические данные в целом удовлетворительно соответствуют результатам расчетов ОКР и указывают на значительные искажения в размерных распределениях по данным СТМ.
По имеющимся изображениям неоплавленных фрагментов (нижняя часть рис. 5б) можно оценить верхнюю границу размера частиц в ЭОРd26: около 15 нм. Независимые опыты проводились с образцами Рd26 большой толщины, для которых рентгенографические исследования затруднялись слабой адгезией наружных слоев осадка. Для отделения тонких фрагментов слабая адгезия оказалась, напротив, благоприятна, и удалось визуализировать достаточно большое число частиц с признаками огранки и размерами 7.5 – 9.5 нм. Разумеется, строение наружных слоев осадков толщиной несколько мкм может отличаться от строения прилегающих к подложке и промежуточных слоев компактных осадков, однако полученные микроскопические данные в целом удовлетворительно соответствуют результатам расчетов ОКР и указывают на значительные искажения в размерных распределениях по данным СТМ.
![]() Более высокие ОКР, полученные в [17], следует объяснить, по-видимому, использованием в эксперименте осадков значительно больших толщин (5 – 15 мкм).
Более высокие ОКР, полученные в [17], следует объяснить, по-видимому, использованием в эксперименте осадков значительно больших толщин (5 – 15 мкм).
![]() Представленные выше данные свидетельствуют о том, что электролитические осадки палладия, полученные в условиях параллельного образования концентрированного гидрида, уникальны не только по сорбционному поведению, но и по объемной и приповерхностной структуре, а также по устойчивости к окислению.
Представленные выше данные свидетельствуют о том, что электролитические осадки палладия, полученные в условиях параллельного образования концентрированного гидрида, уникальны не только по сорбционному поведению, но и по объемной и приповерхностной структуре, а также по устойчивости к окислению.
![]() Обнаруженные особенности дефектности Рd26 следует в целом интерпретировать как сосуществование дефектов с областью действия, ограниченной единицами межплоскостных расстояний в к.к. Рd, и протяженных дефектных областей (возможно, краевых или зернограничных дислокации) с преобладанием первых. Имеющиеся на сегодняшний день данные недостаточны для того чтобы судить о взаимном расположении таких фрагментов разного типа (например, о применимости к Рd26 моделей типа “нанокристаллы в матрице”, которая конкретизирована для Pd в [31]. Однако имеющаяся неопределенность не накладывает ограничений на выбор подхода к термодинамическому описанию свойств аномальных гидридов: полученные структурные данные обосновывают справедливость подхода к аддитивному описанию изотерм сорбции водорода в b-фазе [13], поскольку подтверждают пространственное разделение структурно-различных областей. Отсутствие равномерного распределения дефектов по всему объему Рd26 накладывает некоторые ограничения на самосогласованное описание процессов сорбции водорода этим материалом “единой” изотермой [32].
Обнаруженные особенности дефектности Рd26 следует в целом интерпретировать как сосуществование дефектов с областью действия, ограниченной единицами межплоскостных расстояний в к.к. Рd, и протяженных дефектных областей (возможно, краевых или зернограничных дислокации) с преобладанием первых. Имеющиеся на сегодняшний день данные недостаточны для того чтобы судить о взаимном расположении таких фрагментов разного типа (например, о применимости к Рd26 моделей типа “нанокристаллы в матрице”, которая конкретизирована для Pd в [31]. Однако имеющаяся неопределенность не накладывает ограничений на выбор подхода к термодинамическому описанию свойств аномальных гидридов: полученные структурные данные обосновывают справедливость подхода к аддитивному описанию изотерм сорбции водорода в b-фазе [13], поскольку подтверждают пространственное разделение структурно-различных областей. Отсутствие равномерного распределения дефектов по всему объему Рd26 накладывает некоторые ограничения на самосогласованное описание процессов сорбции водорода этим материалом “единой” изотермой [32].
![]() Согласно [33], избыток водорода в дефектной металлической матрице (по сравнению с равновесной сорбционной емкостью упорядоченной матрицы) может быть обусловлен несколькими причинами:
Согласно [33], избыток водорода в дефектной металлической матрице (по сравнению с равновесной сорбционной емкостью упорядоченной матрицы) может быть обусловлен несколькими причинами:
![]() Полученные в этой работе результаты не позволяют связать особые сорбционные свойства ЭОРd26 только с повышенной концентрацией вакансий или даже говорить о том, что вклад вакансий в случае образования аномальной b-фазы гидрида является существенным. Представляется более вероятным, что вакансии могут быть ответственными за избыточное количество водорода в a-фазе, которое отмечено для всех исследованных выше образцов.
Полученные в этой работе результаты не позволяют связать особые сорбционные свойства ЭОРd26 только с повышенной концентрацией вакансий или даже говорить о том, что вклад вакансий в случае образования аномальной b-фазы гидрида является существенным. Представляется более вероятным, что вакансии могут быть ответственными за избыточное количество водорода в a-фазе, которое отмечено для всех исследованных выше образцов.
![]() Дальнейший прогресс в структурных исследованиях аномальных электролитических осадков палладия может быть связан с использованием методов малоуглового рассеяния [34] и непосредственным изучением концентрированного гидрида, образующегося в дефектной палладиевой матрице [35].
Дальнейший прогресс в структурных исследованиях аномальных электролитических осадков палладия может быть связан с использованием методов малоуглового рассеяния [34] и непосредственным изучением концентрированного гидрида, образующегося в дефектной палладиевой матрице [35].
![]() Работа выполнена при частичной поддержке грантом РФФИ 01-03-33132.
Работа выполнена при частичной поддержке грантом РФФИ 01-03-33132.
Таблица. Периоды элементарной ячейки а Pd , размер кристаллита (области когерентного рассеяния) D и микронапряжения e в различных Pd материалах
| Pd материалы | а (нм) | hkl | D (нм) | e*10-3 |
| Рd26 | 0,38913±0,00020 | 111 200 420 |
||
| Рd80 | 0,38883±0,00014 | 111 200 420 |
||
| Рd120 | 0,38820±0,00012 | 111 200 420 |
||
| Еd = -50 мВ [17] | 0,3884 | 111 | 30 | 1,8 |
| Еd = 250 мВ[17] | 0,3885 | 111 | 70 | 1,5 |
| Н.к. Pd (компакт) [3] | 0,38902±0,00008 | 111, 222 200, 400 220, 440 |
16±2 9±2 13±4 |
2,4±1,5 6±4 5±4 |
| Н.к. Pd (компакт) [10] | 0,38952±0,00015 | 111, 222 200, 400 220, 440 |
14±2 16± 10 5 15± 6 3 |
1,2±1,1 13±4 7±3 |
| К.к. Pd [4] | 0,38914±0,00002 | среднее изотропное значение | 230±20 | 3,0±0,1 |
| К.к. Pd [*] | 3,8886±0,0001 |
![]() Примечание. Измерение периода а н.к. Pd проведено в [4] с большей точностью по сравнению с достигавшейся в [10], т.к использованное в [4] синхротронное излучение (l =0,007848 нм) позволяет регистрировать большее число рефлексов; оно также более интенсивно по сравнению с рентгеновским Cu Ka (Ni фильтр)-излучением, примененным в [10, *], определение же вкладов ОКР и микронапряжений в уширение рефлексов при синхротронном источнике приводит к большей ошибке, нежели при рентгеновском, в связи с большей неопределенностью формы профиля линий в первом случае.
Примечание. Измерение периода а н.к. Pd проведено в [4] с большей точностью по сравнению с достигавшейся в [10], т.к использованное в [4] синхротронное излучение (l =0,007848 нм) позволяет регистрировать большее число рефлексов; оно также более интенсивно по сравнению с рентгеновским Cu Ka (Ni фильтр)-излучением, примененным в [10, *], определение же вкладов ОКР и микронапряжений в уширение рефлексов при синхротронном источнике приводит к большей ошибке, нежели при рентгеновском, в связи с большей неопределенностью формы профиля линий в первом случае.
![]() * Lawson A.C., Conant J.V., Robertson R., Rohwer R.K., Young V.A., Talcott C.L. //J. Alloys Compounds. 1992. V.183. P.174.
* Lawson A.C., Conant J.V., Robertson R., Rohwer R.K., Young V.A., Talcott C.L. //J. Alloys Compounds. 1992. V.183. P.174.